Laser‐Driven Single‐Step Synthesis of Monolithic Prelithiated Silicon‐Graphene Anodes for Ultrahigh‐Performance Zero‐Decay Lithium‐Ion Batteries
Avinash Kothuru, Gil Daffan, Fernando Patolsky*
Nano-Micro Letters (2026)18: 220
https://doi.org/10.1007/s40820-026-02074-2
本文亮点
1. 报道了一种在环境条件下单步激光驱动的工艺,该工艺利用简单前驱体同时合成预锂化硅纳米颗粒并将其整合到坚固的石墨烯基质中。
2. 在酚醛树脂超快光热石墨化过程中,硅(Si)与常见锂盐前驱体之间发生界面固相反应,原位实现了预锂化。
3. 与未锂化对应物相比,预锂化硅纳米颗粒/激光诱导石墨烯负极在锂离子半电池和全电池中展现出卓越的循环稳定性(2000次循环后容量保持率>98%)及近乎零的性能衰减。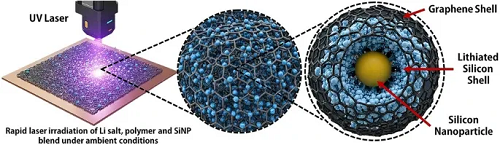
研究背景
锂离子电池(LIBs)为便携式电子设备、电动汽车及电网储能等各类技术提供动力。其高能量密度、长循环寿命和低自放电特性使其成为众多应用场景的首选。随着储能需求的增长,提升锂离子电池能量密度、加快充电速度并延长使用寿命至关重要。在此发展过程中,负极材料是关键因素,其中石墨是最常用的负极材料。尽管石墨成本低且稳定性好,但其低理论容量(372 mAh g⁻¹)限制了能量密度和性能的进一步提升。
内容简介
由于能量密度显著更高,硅基负极为锂离子电池(LIBs)中石墨负极提供了极具前景的替代方案。然而,硅在嵌锂过程中会发生显著的体积膨胀,导致结构不稳定和固体电解质界面(SEI)持续形成,大幅降低初始库仑效率(ICE)和容量保持率,从而限制了其实际应用。采用硅纳米结构化以及与导电碳基质复合等策略虽有助于缓解体积变化并提高导电性,但无法完全解决锂损失和长期容量衰减问题。预锂化可通过补偿锂损失和稳定SEI来缓解这些问题。然而,传统预锂化方法复杂、对空气敏感、需多步且为非原位操作,通常需要使用活性锂金属或特殊锂盐前驱体。为此,以色列特拉维夫大学Fernando Patolsky等人引入了一种激光驱动的固态下原位预锂化方法,该方法与硅-石墨烯类单块复合负极的合成同步进行。将酚醛树脂、硅纳米颗粒(SiNPs)和常见锂盐的三元混合物置于快速低功率激光辐照下,可制备出自支撑、空气稳定的预锂化复合材料,其中形成的多孔导电基质包裹着SiNPs,而独特的激光诱导环境会触发原位反应,对硅表面进行预锂化并形成稳定的共价界面。所得锂化负极展现出卓越性能,在5 A g⁻¹下循环2000次以上容量衰减可忽略不计(<2%),比容量超过1700 mAh g⁻¹,4500次循环后容量保持率为83%,且初始库仑效率高于97%(与未锂化负极相比)。该负极还具备超快充能力,在10 A g⁻¹下可保留高达63%的最大容量。这一创新不仅推动了下一代锂离子电池的开发,还为将易获取且成本低廉的前驱体材料转化为高性能电极建立了框架,有望降低电池制造的复杂性和成本。
图文导读
I 合成方法和机理概述
图1a展示了合成过程的示意图。制备了由无水乙醇和酚醛树脂组成的溶液。选择酚醛树脂作为碳前驱体,是因为它是一种用于激光诱导石墨烯(LIG)形成的成熟材料,能够在能量存储器件中生成具有优异机械稳定性和电化学性能的高导电性且均匀的石墨烯薄膜。图1b展示了激光辐照期间前驱体结构内拟发生的分子尺度反应机制,后续将通过严格的结构和形貌表征进一步证实。图1c展示了涂有前驱体浆料的大尺寸(长度20 cm)铜箔以及经激光处理后的产物,凸显了该方法的可扩展性。
图1. a 对锂盐、酚醛树脂和硅纳米颗粒(SiNPs)混合物应用单步、环境条件、低功率激光辐照过程的示意图,该过程用于合成自支撑、多孔、预锂化的PL-SiNP/激光诱导石墨烯(LIG)复合负极。b 三元混合物在激光辐照下的分子尺度机制示意图,该机制在诱导LIG形成的同时,同步触发硅纳米颗粒的原位预锂化和包覆。c 展示大尺寸片材形成的预锂化硅纳米颗粒/激光诱导石墨烯负极合成过程,凸显该工艺的可扩展性。
II 结构和形态分析
如图2a、b所示,采用高分辨率扫描电子显微镜(HR-SEM)对激光辐照后非预锂化和预锂化(PL)SiNP/LIG复合材料的结构和形貌特征进行了研究,同时展示了相应的能量色散X射线光谱(EDS)元素分布图。值得注意的是,EDS分布图中不存在Li,这是因为Li的原子序数较低,在与电子束相互作用时不发射强特征X射线信号。因此,为了在微观尺度上准确确定LIG基质中Li和Si分布的均匀性,对样品进行了飞行时间二次离子质谱(TOF-SIMS)分析,如图2c所示。为了进一步阐明纳米级结构并确认复合材料中Li的分布,进行了扫描透射电子显微镜(STEM)成像,如图2d、e所示。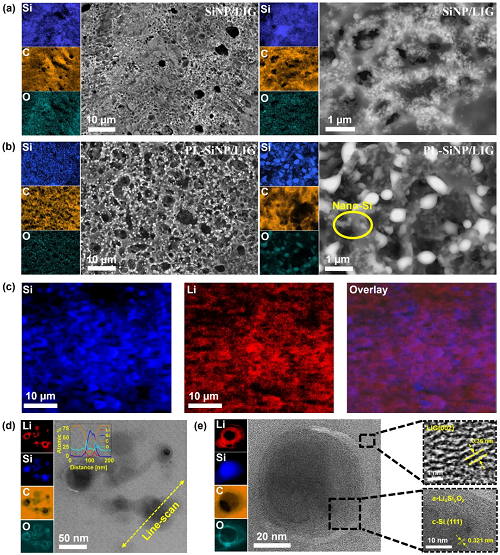
图2. a不同放大倍数下非预锂化SiNP/LIG复合材料的HR-SEM图像及相应EDS分布图,突出了LIG基质内的高孔隙率和均匀的SiNP分布。b原位预锂化(PL)SiNP/LIG复合材料的HR-SEM图像及相应EDS分布图。c微尺度下PL-SiNP/LIG复合材料表面的TOF-SIMS分布图。d LIG基质内预锂化SiNPs的低倍STEM图像,EELS分布图显示存在锂化SiNP壳层。左上角插图显示沿粒子直径的元素分布线扫描EELS谱图。e 预锂化SiNP的高倍STEM图像及EELS分布图。上方插图显示LIG基质的HR-STEM图像及测量的面间距,下方插图显示非晶锂化壳层与晶体Si的界面。
图3a展示了SiNP/LIG和PL-SiNP/LIG复合材料的粉末X射线衍射(PXRD)图谱,对其进行检测旨在识别主要的体相晶体相,并评估其对用于负极的晶体硅活性材料的影响。所有峰均使用无机晶体结构数据库(ICSD)进行鉴定,图中相应给出了参考编号。图3b中,对SiNP/LIG和PL-SiNP/LIG复合材料进行了拉曼光谱分析,以评估石墨烯的质量和结构特征。G带(~1580 cm⁻¹)对应于sp²杂化碳原子的面内振动模式,表明具有石墨结晶度,其强度越高则电导率越大。进行了精细的Ar⁺离子簇溅射,如图3c所示,实现了从表面起数十纳米范围的深度分析。该分析得出Si、Li和O的平均原子百分比分别为11.5%、21.1%和37.6%,表明表面存在硅酸锂相,可能还存在其他含锂化合物。图3d中的Li 1s谱显示,从LiOH前驱体在~54.2 eV处的峰显著偏移至在~55.8 eV处出现的另一个主峰,此前多项研究将此归因于硅酸锂相。图3e中的Si 2p谱展示了原始SiNPs、SiNP/LIG和PL-SiNP/LIG的表面组成。虽然PXRD和拉曼光谱证实,两种复合材料中大部分硅材料仍为元素硅,但表面显然已发生化学转变,如今呈现出与更高氧化态一致的更高结合能相。图3f所示的C 1s谱对酚醛树脂(PR)LIG前驱体、SiNP/LIG和PL-SiNP/LIG样品进行了比较,以分析LIG的化学性质。采用氮气(N₂)获得气体吸附和脱附等温线,并通过Brunauer-Emmett-Teller(BET)方法(图3g)进行分析,以评估SiNP/LIG和PL-SiNP/LIG复合材料的比表面积(SSA)和孔径分布。从图3h中的等温线推导得出的Barrett-Joyner-Halenda(BJH)孔径分布,进一步揭示了材料的孔隙率。发达的多孔结构可促进离子传输,并容纳硅约400%的体积膨胀,从而减轻粉化并在循环过程中保持电极完整性。图3i展示了电感耦合等离子体质谱(ICP-MS)分析,该分析旨在评估激光处理后锂物种发生化学转变对锂离子电池碳酸盐基电解液中锂溶解度的影响。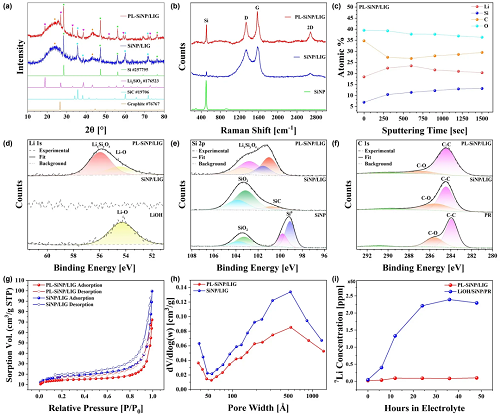
图3. a PL-SiNP/LIG与SiNP/LIG的PXRD图谱及ICSD参考峰。b PL-SiNP/LIG、SiNP/LIG和原始SiNPs的拉曼光谱。c PL-SiNP/LIG中Li、Si、C和O原子百分比的XPS深度剖面。d PL-SiNP/LIG、SiNP/LIG和LiOH前驱体的XPS Li 1s谱。e PL-SiNP/LIG、SiNP/LIG和SiNP前驱体的XPS Si 2p谱。f PL-SiNP/LIG、SiNP/LIG和酚醛树脂(PR)参考样品的XPS C 1s谱。g PL-SiNP/LIG和SiNP/LIG的BET N₂吸附-脱附等温线。h PL-SiNP/LIG和SiNP/LIG的BJH孔径分布。i PL-SiNP/LIG与未辐照的LiOH/SiNP/酚醛树脂前驱体混合物浸入电解液后随时间变化的Li⁺浓度ICP-MS分析。
III 硅锂比对负极性能的影响
采用初始前驱体浆料中不同SiNP:LiOH质量比,对PL-SiNP/LIG负极在半电池中的电化学性能进行了广泛评估,以评估其对离子传输、电荷转移动力学和整体电化学稳定性的影响,如图4所示。图4a展示了负极的恒电位电化学阻抗谱(PEIS)分析结果,等效电路模型如图S3所示。图4b总结了图4a和图S4的研究结果,展示了从瓦尔堡常数中提取的锂离子扩散系数。相应地,图4c展示了在不同Si:LiOH质量比下PL-SiNP/LIG负极的倍率性能分析结果,该分析旨在评估预锂化SiNPs和锂储存库在不同电流密度下对充放电行为的影响。图4d展示了在高电流密度5 A g⁻¹下PL-SiNP/LIG负极的恒电流循环性能,评估了前驱体混合物中不同Si:LiOH比例下,初始库仑效率(ICE)、质量比容量和1100次循环后的长期稳定性。图4e展示了1:1和1:0两种成分在2000次以上循环中的扩展循环数据。1:1预锂化负极的容量保持率为98.5%,而1:0非预锂化负极的容量保持率仅为60.1%。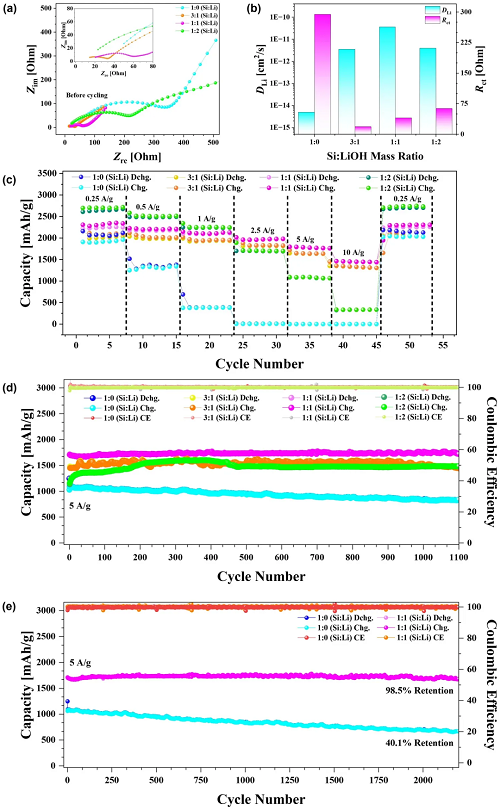
图4. a 循环前对不同Si:LiOH质量比的PL-SiNP/LIG复合材料进行恒电位电化学阻抗谱(PEIS)分析。b 从电化学阻抗谱(EIS)图中提取的不同Si:LiOH比例下PL-SiNP/LIG负极的锂离子扩散系数和电荷转移电阻。c 不同Si:LiOH比例下PL-SiNP/LIG负极的倍率性能。d 不同Si:LiOH比例的PL-SiNP/LIG负极在5 A g⁻¹下进行1100次恒电流循环。e 对d中1:1和1:0样品继续进行恒电流循环,循环次数达2000次以上。
IV 锂盐前驱体对负极性能的影响
为进一步探究所提出预锂化方法的普适性及其与常见锂盐前驱体的兼容性,有必要评估除LiOH之外的其他替代物。虽然LiOH已展现出卓越的电化学性能,但研究其他锂盐有助于深入了解该方法的更广泛适用性。为此,评估了Li₂CO₃、LiNO₃、LiF和LiClO₄作为替代预锂化源的情况。如图5所示,系统分析了它们对锂离子电池(LIBs)电化学性能的影响。图5a展示了循环前进行的恒电位电化学阻抗谱(PEIS)分析结果,表明所有锂盐的阻抗行为总体相似,但存在细微差异。图S19通过瓦尔堡斜率进一步研究了这些差异,结果显示所有成分的DLi相似。其中,LiOH的斜率最低,对应最高的DLi,证实了其优异的锂离子传输性能。图5b总结了从电化学阻抗谱(EIS)拟合中提取的DLi和Rct。图5c进一步支持了扩散和电荷转移能力方面观察到的趋势,表明LiOH在不同倍率下既能提供最高容量,又能实现最佳容量保持率,进一步巩固了其卓越的电化学性能。图5d进一步凸显了添加锂盐的普遍益处,展示了在5 A g⁻¹的高电流密度下,经过140多次循环仍具有出色的循环稳定性。除增强长期性能和结构耐久性外,结果还证实了倍率性能的提升,即使在快速充电条件下也能保持稳定容量。LiOH产生的强碱性条件(共轭酸H₂O,pKa ≥ 14)能够使树脂中的酚羟基(pKa ~ 10)和SiNPs表面上的硅醇(Si–OH)基团(pKa ~ 4–7)有效去质子化,形成带负电荷的位点,吸引Li+离子,并促进前驱体基质内的界面致密化,如图5e所示的示意图所示。 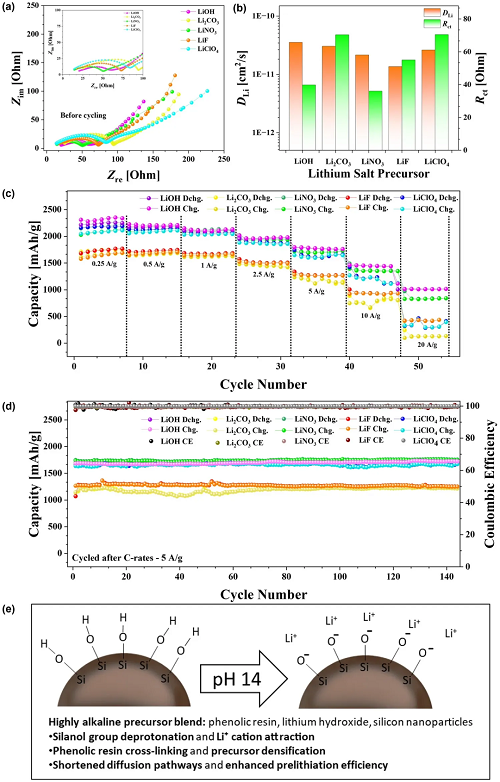
图5. a 循环前对采用不同锂前驱体的PL-SiNP/LIG复合材料进行恒电位电化学阻抗谱(PEIS)分析,等效电路模型如图S3所示。b 采用不同锂前驱体的PL-SiNP/LIG负极的锂离子扩散系数(DLi)和电荷转移电阻(Rct)。c 采用不同锂前驱体的PL-SiNP/LIG负极的倍率性能。d 采用不同锂前驱体的PL-SiNP/LIG负极在5 A g⁻¹下的恒电流循环,在倍率性能分析后进行循环测试。e 示意图展示了氢氧化锂在碱性前驱体混合物中促进硅醇去质子化、吸引Li+、树脂交联以及提高反应效率的作用。
V 失效分析
图6对经过100次循环后的SiNP/LIG和PL-SiNP/LIG负极进行了对比性的失效后分析,突出了SEI形成和结构完整性方面的关键差异。该分析对于评估预锂化在缓解硅基负极典型降解途径方面的有效性至关重要。图6a展示了未预锂化的SiNP/LIG负极SEM图像及对应的EDS面扫图。图6b展示了同一未预锂化样品的扫描透射电子显微镜(STEM)和电子能量损失谱(EELS)面扫图,用于考察锂的分布情况。未观察到硅和锂信号之间存在空间相关性,且SEI内的锂含量似乎较低。相比之下,图6c、d展示了经过100次循环后的预锂化PL-SiNP/LIG负极的失效后分析结果,显示出显著的结构保留情况。多孔结构基本保持完整,与循环前的状态极为相似。图6d通过失效后预锂化负极的STEM/EELS锂面扫图进一步支持了这一结论,该图揭示了锂和硅信号之间存在显著的空间重叠。图6e展示了两种负极的C 1s光谱。观察到两个主要峰:一个在约284.4 eV处,对应于C–C/C=C键(主要来自底层的激光诱导石墨烯(LIG));另一个在约290 eV处,归因于C–F键,这是SEI中含氟有机成分的一部分。图6f展示了F 1s光谱。未预锂化样品在约685.5 eV处呈现出一个较弱的氟化锂(LiF)峰,并伴有在约687.5 eV处一个相对较强的峰,该峰对应于SEI内含氟碳物种中的C–F键。图6g和图S22分别展示了Si 2p和Li 1s光谱。虽然预锂化和未预锂化样品在循环前后峰的位置和形状大致相似,但与信息更丰富的F 1s和C 1s光谱相比,这些光谱提供的额外信息有限。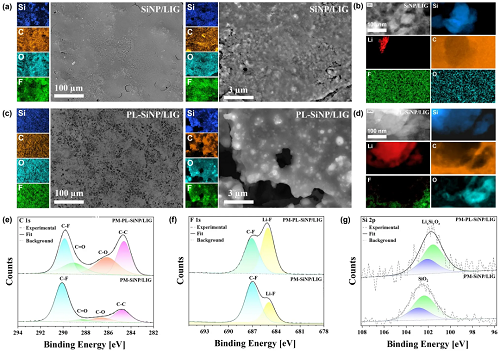
图6. 循环100次后的失效后分析。a 失效后未预锂化负极的扫描电子显微镜/能量色散X射线光谱(SEM/EDS)分析。b 失效后未预锂化负极的扫描透射电子显微镜/电子能量损失谱(STEM/EELS)面扫分析。c 失效后预锂化负极的SEM/EDS分析。d 失效后预锂化负极的STEM/EELS面扫分析。e 失效后预锂化与未预锂化负极的X射线光电子能谱(XPS)C 1s光谱。f 循环100次后失效后两种负极的XPS F 1s光谱。g 循环100次后失效后两种负极的XPS Si 2p光谱。
VI 总结与展望
本研究首次展示了针对硅-石墨烯复合负极的一种基于激光的、在常温常压下进行的固态原位预锂化技术。通过利用激光诱导石墨烯(LIG)形成过程中产生的极端局部热和压力条件,这种单步方法能够在大气环境下同时实现多孔石墨烯基质的合成、硅纳米颗粒(SiNP)的包覆以及预锂化过程,无需使用粘结剂、导电添加剂、有害锂源或受控环境。所得到的预锂化负极展现出卓越的电化学性能,包括高初始库仑效率(ICE)、增强的锂离子扩散能力,以及在高电流密度下经过2000多次循环后容量衰减接近于零,其性能优于未预锂化的同类产品以及其他已报道的纳米硅负极。
作者简介

关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2024 JCR IF=36.3,学科排名Q1区前2%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 以色列特拉维夫大学Fernando Patolsky等:超高性能零衰减锂离子电池单片预锂化硅石墨烯负极的激光驱动一步合成