研究背景
无水锂-氧电池(LOBs)因其超高的理论能量密度(3,500 W h kg⁻1),被认为是取代锂离子电池(LIBs)用于电动汽车、机器人和大规模电网的最具潜力的候选体系之一。通常,其在循环过程中的反应机制基于 Li⁺ + O₂ ↔ Li₂O₂(E⁰ = 2.96 V vs. Li/Li⁺),其中放电涉及氧还原反应(ORR),充电涉及氧析出反应(OER)。然而,如此强大的体系在进一步应用中受到较差的倍率性能、有限的循环寿命以及高过电位的严重阻碍。这些问题主要源于其迟缓的氧化还原动力学,导致放电产物(Li₂O₂)以及甚至绝缘的副反应产物(LiOH 和 Li₂CO₃)在正极上的堆积。鉴于正极应暴露三相接触界面区域(正极/电解质/O₂)并能够存储放电产物,采用具备快速电子传输速率和可逆放电产物形成能力的理想正极催化剂,将大幅提升其电催化性能,并加速 LOBs 的商业化应用进程。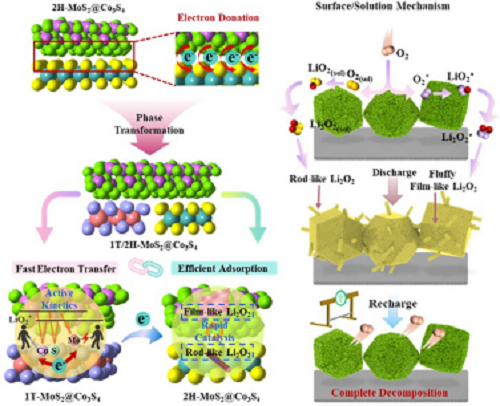
Constructing Double Heterojunctions on 1T/2H-MoS₂@Co₃S₄ Electrocatalysts for Regulating Li₂O₂ Formation in Lithium‐Oxygen Batteries
Yichuan Dou, Zhuang Liu, Lanling Zhao*, Jian Zhang, Fanpeng Meng, Yao Liu, Zidong Zhang, Xingao Li, Zheng Shang, Lu Wang, Jun Wang*
Nano-Micro Letters (2026)18: 51
https://doi.org/10.1007/s40820-025-01895-x
本文亮点
1. 通过Co向Mo原子的界面提供电荷构筑了1T/2H-MoS₂@Co₃S₄电催化剂,形成了包括 1T-MoS₂@Co₃S₄ 和 2H-MoS₂@Co₃S₄ 在内的双异质结。
2. 双异质结的互补效应不仅在Co–S–Mo耦合位点触发了快速电荷传输,还使eg轨道保持适度占据,用于吸附含氧中间体以实现高效氧电催化。
3. 获得了溶液路径和表面路径双反应机制的最优吸附能,在循环过程中形成两种放电产物形貌,从而提升 Li–O₂ 电池的性能。
内容简介
Co₃S₄电催化剂因其具有混合价态的Co离子以及优异的结构稳定性而表现出良好的氧析出反应(OER)活性,但由于其电导率较差以及对中间体吸附过强,仍难以用于构建可充电锂氧电池(LOBs)。在本工作中,山东大学王俊、赵兰玲等人通过 Co 向 Mo 离子的电荷供给构筑了 1T/2H-MoS₂@Co₃S₄(1T/2H-MCS)上的精巧双异质结,从而诱导 MoS₂ 由 2H 相向 1T 相转变。这些双异质结的独特特征赋予 1T/2H-MCS 在充放电过程中具有互补催化能力。值得注意的是,1T-MoS₂@Co₃S₄ 可提供快速的 Co–S–Mo 电子传输通道以促进 ORR/OER 动力学,而 2H-MoS₂@Co₃S₄ 有助于在吸附含氧中间体时实现 eg 轨道占据。在此基础上,Li₂O₂ 的成核路径转变为溶液与表面双路径机制,从而改善其可逆沉积与分解动力学。最终,1T/2H-MCS 正极相比 Co₃S₄ 和 MoS₂ 正极表现出更优的电催化性能。这一创新的异质结构设计为通过提升电导率和调控对含氧中间体的吸附来构建高效过渡金属硫化物催化剂用于 LOBs 提供了一种可靠策略。
图文导读
I 1T/2H-MoS₂@Co₃S₄ 中双重互补效应的理论预测
通过 Bader 电荷分析表明,电子以 0.638 e 的量从 Co₃S₄ 转移至 2H-MoS₂,诱导 2H-MoS₂ 向 1T-MoS₂ 相的转变(图 1a)。电荷密度差和电子局域函数(ELF)进一步证明了电子在异质界面上从 Co₃S₄ 转移至 2H-MoS₂,如图 1b 所示。图 1c 中,当 2H-MoS₂ 与 Co₃S₄ 结合时,Co₃S₄ 赠予的额外电子首先占据 2H-MoS₂ 的 e′ 轨道,使其结构不稳定并诱导 Mo 4d 轨道重组,从而使 MoS₂ 由 2H 相转变为 1T 相。结果,MoS₂ 的固有电导率显著提高,可作为 Co₃S₄ 的导电相。图 1d 中费米能级附近的总态密度(TDOS)显示 1T-MoS₂@Co₃S₄ 具有更强的金属性。图 1e 中 Co 3d、S 2p 和 Mo 4d 轨道显示显著重叠,表明它们之间存在强电子相互作用,从而加速 Co–S–Mo 通道中的电子传输。此外,在 MoS₂ 与 Co₃S₄ 结合后,从 Co 转移至 Mo 离子的电子引起 Co 的 Ed 下移,而 Mo 则呈相反趋势(图 1f),从而缩小了 Co 3d 与 S 2p 能带中心以及 Mo 4d 与 S 2p 能带中心之间的能量差,并导致 Co、S 和 Mo 之间的强杂化(图 1g)。基于上述分析,图 1h 系统地展示了 1T/2H-MCS 中由 Co 向 Mo 离子电荷馈赠产生的双重互补异质结的协同效应。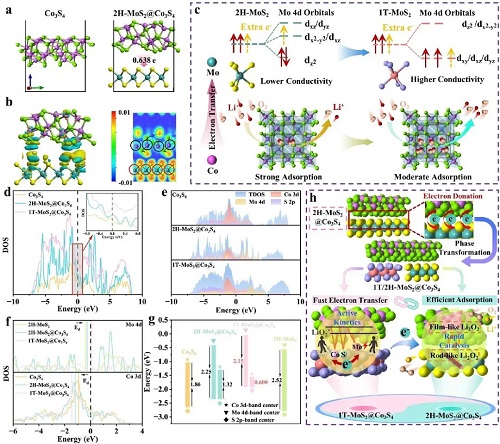
图1. a 2H-MoS₂@Co₃S₄ 的 Bader 电荷转移,b ELF 及电荷密度差,其中电荷积累和耗尽分别以黄色和青色表示。c Co₃S₄ 与 2H-MoS₂ 之间拟议电子转移过程示意图。d 不同样品的 TDOS,e 不同样品的 PDOS。f、g Co 3d、Mo 4d 和 S 2p 的 d 带与 p 带中心。h 双异质结对 1T/2H-MCS 在 LOBs 中作用的示意图。
II 形态和结构表征
在典型的合成过程中,采用 ZIF-67 作为模板,通过简便的水热法制备 1T/2H-MCS(图 2a),该过程可分为两步。为了探索不同样品的晶体结构和组成,进行了 XRD、Raman、XPS 及 EPR 测量。图 2b 的 XRD 分析显示,所有特征峰均对应于立方相 Co₃S₄(JCPDS 47-1738)和 2H-MoS₂(JCPDS 75-1539),证明 1T/2H-MCS 已成功合成。为了提供直接对比,图 2e 展示了 Co₃S₄ 与 1T/2H-MCS 的 Co 2p XPS 谱。在 Mo 3d XPS 谱的解卷积结果中,MoS₂ 中 1T 相的比例超过 65%(图 2d)。因此,Co3⁺ 含量的增加可诱导额外电子,占据 2H-MoS₂ 的 e′ 轨道,从而实现 Mo 4d 轨道重组及 MoS₂ 由 2H 相向 1T 相的转变。为了进一步探测 1T/2H-MCS 中 Co 物种的电子结构及配位信息,进行了 X 射线吸收光谱(XAS)分析。
图 2e 的 Co K 边 X 射线近边吸收光谱(XANES)显示,1T/2H-MCS 的 Co K 边相较于 Co 金属箔和 Co₃S₄ 向高能位置偏移,表明 1T/2H-MCS 中 Co 的化学态升高。1T/2H-MCS 中较高的 Co 价态与理论预测及 XPS 结果一致,提示在与 2H-MoS₂ 结合时,由于电负性差异,电子从 Co 转移至 Mo 位点。图 2f 所示 1T/2H-MCS 的傅里叶变换(FT)k3 加权 EXAFS 谱显示两个明显峰位于 1.63 Å 和 2.52 Å,对应于 Co–S 和 Co–Co。与 Co₃S₄ 相比,1T/2H-MCS 的第一配位路径延长,可归因于与 2H-MoS₂ 结合后 Co–S–Mo 的不对称结构。在图 2g 中,通过 EXAFS 小波变换(WT)中的最大值可更清晰地观察到此配位路径,其与 Co 金属箔和 Co₃S₄ 明显不同。图 2h 为 1T/2H-MCS 的 FESEM 图像,其保留了 ZIF-67 的典型十二面体菱形形貌,并附着随机纳米片,平均颗粒尺寸约 500 nm。如图 2i所示,高角环形暗场(HAADF)及高分辨透射电子显微镜(HRTEM)图像证实 1T/2H-MCS 显示空心结构,包含 Co₃S₄ 纳米多面体及外层 MoS₂ 纳米片。此外,在 MoS₂ 与 Co₃S₄ 异质结构形成后,图 2j 中同时观察到蓝色区域的 1T-MoS₂ 和绿色区域的 2H-MoS₂ 晶格结构,进一步证实了 1T/2H-MCS 的成功合成。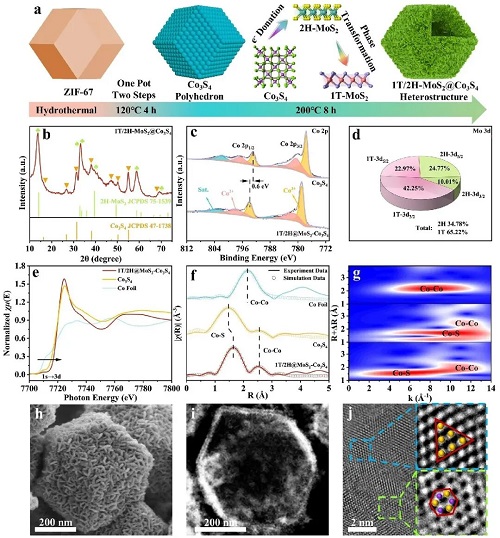
图2. a 1T/2H-MCS 合成过程示意图及拟议的相变机制。b XRD 图谱,c 高分辨 Co 2p XPS 谱,e Co K 边 XANES 谱,f Co K 边 EXAFS 谱的傅里叶变换,g 不同样品的小波变换分析。d 1T 相与 2H 相比例,h FESEM 图像,i HAADF 图像,j HRTEM 图像及对应选区的放大结果(1T/2H-MCS)。
III 电催化性能
为了深入了解所制备催化剂在 LOBs 中的 ORR/OER 催化活性,进行了电化学测试。图 3a 显示了不同正极在 2.35–4.50 V 电压范围内的 CV 曲线。图 3b 展示了不同正极在 100 mA g⁻1 下的初始恒流放电/充电性能。结果显示,1T/2H-MCS 正极表现出最高的放电/充电比容量,分别为 18,721/18,313 mAh g⁻1,而 Co₃S₄、2H-MoS₂ 和 KB 正极的放电/充电比容量分别为 12,823/10,775、8,346/7,744 和 5,195/3,394 mAh g⁻1。图 3c 中,1T/2H-MCS 正极的放电/充电极化随电流密度增加而逐渐上升,在 200、500、800 和 1,000 mA g⁻1 下,仍分别可提供 17,452、12,804、9,852 和 5,203 mAh g⁻1 的放电比容量,对应的库伦效率分别为 97.2%、91.3%、86.7% 和 68.9%。
研究了不同正极在 LOBs 中的长期循环性能。1T/2H-MCS 正极可稳定工作 344 个循环,而 2H-MoS₂、Co₃S₄ 和 KB 正极在 500 mA g⁻1 下以 600 mAh g⁻1 截止比容量分别在 176、149 和 60 个循环后逐渐衰减。图 3d–f 也显示,1T/2H-MCS 正极在 1000 mA g⁻1 的高电流密度下表现出 535 个循环的优异循环寿命,并在固定比容量 1,000 mAh g⁻1 下实现超长 2,420 h 的循环寿命。图 3g 及表 S3 对比了本文结果与包含 Mo、Co 及贵金属化合物的代表性及最先进正极的电池性能。值得注意的是,1T/2H-MCS 正极的循环稳定性表现出可比性能,甚至优于部分贵金属基正极。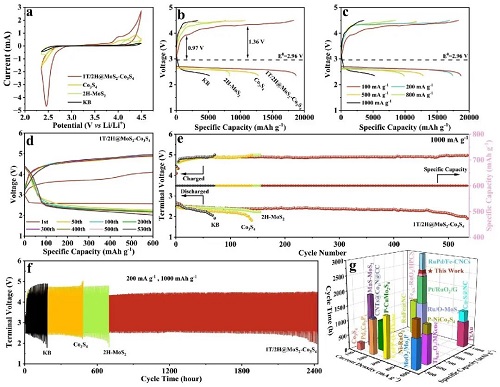
图3. a 不同正极的 CV 曲线,b 在 100 mA g⁻1 下的初始放电/充电曲线,c 速率性能,以及 e 在 1000 mA g⁻1 下、比容量限制为 600 mAh g⁻1 条件下的循环寿命,d 对应的选定充放电曲线,f 在 200 mA g⁻1 下、比容量限制为 1,000 mAh g⁻1 的循环性能。g 1T/2H-MCS 正极与包含 Mo、Co 及贵金属化合物的代表性及最先进正极的循环稳定性对比。
IV 软包电池中的实际应用
图4a表明构建了柔性软包型锂氧电池(LOB)。如图4b所示,其可获得16,875/16252 mAh g⁻1的放电/充电比容量以及96%的高容量保持率,表1T/2H-MCS正极具有优异的ORR/OER动力学。令人印象深刻的是,在0.5 mA cm⁻2的电流密度下,以1 mAh cm⁻2的限定比容量可连续运行375个循环,展现出相当可观的循环稳定性,如图4c所示。为清晰展示电池的电化学稳定性,对采用1T/2H-MCS正极的锂氧电池在不同弯曲角度下的开路电压进行了测试(图4d),结果显示即使电池一角被剪切且同时处于180°弯曲状态,开路电压也几乎无变化。此外还表明,两个软包型锂氧电池串联可获得5.84 V的开路电压。如图4e所示,上述电池可成功为智能手机充电,还可为智能手表充电,21 min内其电量从73%增加至84%(图4f)。这表明采用1T/2H-MCS正极的软包型锂氧电池具有高能量密度和耐久性,对支持下一代电子设备极为有利。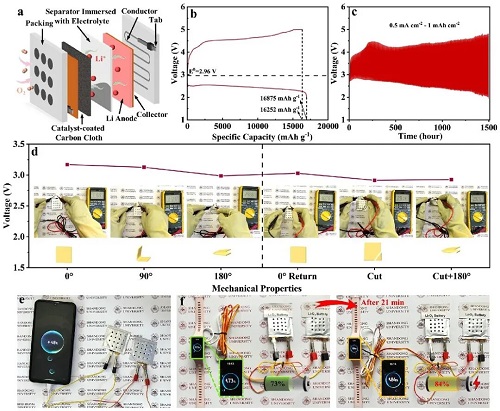
图4. a 软包LOBs结构的示意图;b 初始放电/充电性能;c 在1800 mAh g⁻1的特定比容量限制下,于600 mA g⁻1电流密度下的循环时长;d 不同变形状态下的开路电压;e、f 软包型锂氧电池的实际应用。
V 电化学分析
从非原位FESEM图像中观察到1T/2H-MCS、Co₃S₄和2H-MoS₂正极在第1次放电/充电过程中不同阶段的形貌演变。如图5a、b所示,显然致密的膜状放电产物附着在Co₃S₄正极表面,导致正极钝化且比容量受限。此外,1T/2H-MCS正极在第100次充电后仍能保持原始形貌。相比之下,如图5c、f所示,Co₃S₄和2H-MoS₂正极表面仍残留大量物质。为进一步探究放电产物的化学状态及放电/充电过程中的反应机理,研究了在200 mA g⁻1电流密度下,具有选定关键状态(图5g、h)的1T/2H-MCS正极的非原位高分辨率Li 1s XPS。在充电过程中,于500 mA g⁻1电流密度下获得了不同正极的DEMS结果,如图5i所示。显然,1T/2H-MCS的e⁻/O₂比值接近1.92,表明Li₂O₂ (2e⁻/O₂)和LiO₂ (e⁻/O₂)同时发生分解。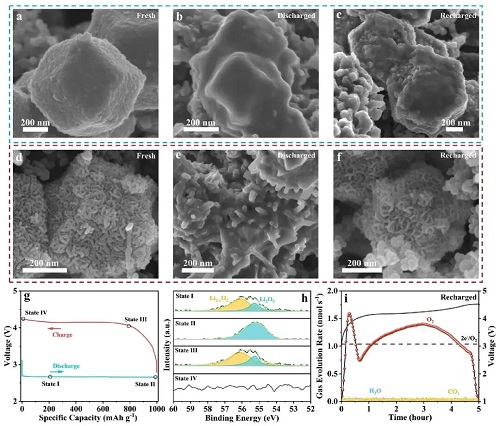
图5. a – c为Co₃S₄正极在不同状态下的FESEM图像,d – f为1T/2H-MCS正极在不同状态下的FESEM图像。g为在200 mA g⁻1电流密度下,截止比容量为1000 mAh g⁻1时的初始放电/充电曲线,h为不同状态下的高分辨率Li 1s XPS图谱,i为1T/2H-MCS正极在200 mA g⁻1电流密度下充电曲线对应的DEMS谱图。
VI 通过DFT计算提出的机制
图S41展示了吸附不同氧物种(LiO₂、Li₂O₄、Li₃O₄和Li₄O₄)后的不同优化结构,Co₃S₄、2H-MoS₂、2H-MoS₂@Co₃S₄和1T-MoS₂@Co₃S₄表面不同氧物种对应的电荷密度差图和Bader电荷转移情况分别如图6a所示。通常,金属-氧键共价性的适度增强倾向于降低电子转移能垒。从图6b中可以发现,与Co₃S₄和1T-MoS₂@Co₃S₄相比,2H-MoS₂@Co₃S₄中Co 3d轨道和O 2p轨道在费米能级附近的重叠程度适中,这表明LiO₂在2H-MoS₂@Co₃S₄上的吸附适度。同时,从图6b中可以观察到Co 3d和O 2p能带中心之间的能隙增大。此外,2H-MoS₂@Co₃S₄具有优化的Co的d带中心值(Ed),且Co – O反键轨道占据适度,这有助于Co位点与含氧中间体之间形成最佳键合相互作用,实现适宜的氧吸附并促进电催化(图6c)。从图S46中可以追踪到吸附LiO₂前后四个模型的明显差异,这表明由于与LiO₂的d – p轨道杂化得到优化,2H-MoS₂@Co₃S₄具有有效的吸附作用。图6d清晰地展示了四种催化剂对反应中间体的详细吸附能。
在图6f中,2H-MoS₂@Co₃S₄正极在Co₃S₄(0.86/2.27 V)、1T-MoS₂@Co₃S₄(1.17/0.81 V)和MoS₂(0.70/1.54 V)中表现出最低的OER过电位/ ORR过电位(0.16/0.18 V),这表明由于与含氧中间体适度的d-p轨道杂化,2H-MoS₂@Co₃S₄对锂-氧电池(LOBs)中氧氧化还原反应的电催化活性显著提高(图6e,g)。基于上述结果,可在图6h中建立含氧中间体与不同催化剂的结合能和过电位之间的火山型关系。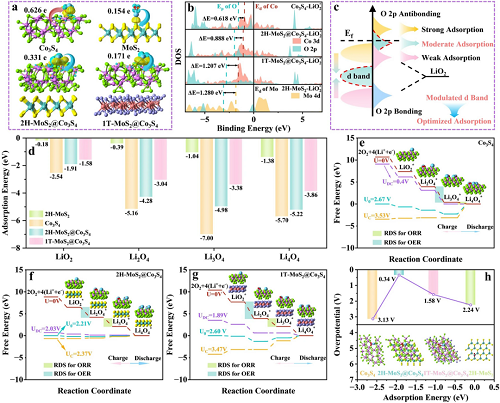
图6. a 不同催化剂的电荷密度差及Bader电荷转移情况;b 不同催化剂中Co 3d的d带中心与S 2p的p带中心;c 1T/2H-MCS的d-p轨道杂化示意图;d 吸附能及吸附能与过电位的关系;e-g 计算得到的能量图;h 不同催化剂吸附能与过电位的关系。
方案1阐明了不同正极上放电产物的合理形成与分解路径。提出在正极表面通过单电子还原反应首先生成中间产物LiO₂。对于2H-MoS₂正极,由于对LiO₂的结合能极弱,该中间产物随后溶解于电解液中,记为LiO₂(sol),并通过溶液生长路径进一步转化为棒状Li₂O₂。在充电过程中,2H-MoS₂正极存在催化活性低、导电性差以及与棒状Li₂O₂接触有限等问题,导致其放电比容量极低且循环性能不佳。相反,LiO₂在Co₃S₄表面具有较强的吸附能,促使Li₂O₂通过表面生长路径形成致密的膜状结构(图5b)。这些膜状Li₂O₂紧密附着于正极表面,减缓了电子/离子在正极催化剂上的动力学扩散,降低了活化程度并导致较高的界面极化。幸运的是,由于1T/2H-MCS对LiO₂具有适中的吸附能,Li₂O₂通过溶液和表面双重路径在1T/2H-MCS正极上生长,形成了两种不同形貌(图5e)。1T/2H-MCS通过1T-MoS₂@Co₃S₄和2H-MoS₂@Co₃S₄双互补异质结的协同效应增强了催化活性,改变了催化剂表面与反应中间体之间的亲和力,从而影响LiO₂的成核过程,成功实现了理想的Li₂O₂形貌。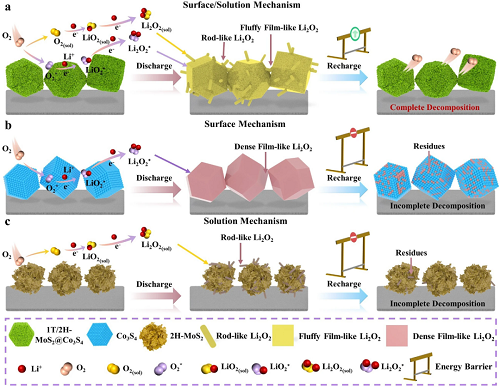
方案1. a 1T/2H-MCS、b Co₃S₄和c 2H-MoS₂正极在ORR/OER过程中的电催化机制。
VII 总结
总之,以ZIF – 67为模板,通过一锅水热法结合两步升温工艺合成了1T/2H – MCS复合材料,该材料可作为LOBs的高效双功能电催化剂。显然,1T/2H – MCS正极展现出非凡的电化学性能,包括在100 mA g⁻1下具有18,721/18,500 mAh g⁻1的高可逆放电/充电比容量,以及在100 mA g⁻1、截止比容量为1000 mAh g⁻1的条件下具有2420 h的超长循环寿命。当应用于软包型锂 – 氧电池时,1T/2H – MCS正极展现出令人印象深刻且具有实际应用潜力的性能。基于DFT计算和实验结果,提出1T/2H – MCS正极显著提升的OER/ORR性能归因于由Co向Mo离子电荷转移产生的双重异质结的互补效应。首先,1T – MoS₂@Co₃S₄提供了Co – S – Mo键无阻碍的电子传输通道,在1T/2H – MCS内很好地重新分配电子,从而显著改善其界面电子转移动力学和电催化活性。然后,2H – MoS₂@Co₃S₄在吸附含氧中间体时具有适中的反键轨道占据数,通过调节吸附能优化放电产物的形貌并加速其分解。此外,1T/2H – MCS继承了ZIF – 67的十二面体空心结构,并装饰有MoS₂纳米片,这可以限制MoS₂的层状取向生长并增加比表面积以暴露大量稳定的活性位点。这种互补关系为设计用于锂 – 氧电池的具有双重异质结的高效电催化剂提供了指导。
作者简介


关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2024 JCR IF=36.3,学科排名Q1区前2%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 山东大学王俊&赵兰玲等:双异质结电催化剂提升锂氧电池性能
 苏州大学汪晓巧/张克勤等:微流控纺丝构筑高性能PEDOT:PSS热电非织造布
苏州大学汪晓巧/张克勤等:微流控纺丝构筑高性能PEDOT:PSS热电非织造布 合肥工大沈王强&华科卢兴等:金属单原子协同钴团簇负载于纳米碳骨架实现高效过氧化氢电合成
合肥工大沈王强&华科卢兴等:金属单原子协同钴团簇负载于纳米碳骨架实现高效过氧化氢电合成 暨南大学王有生/范建东/麦耀华等:超44%宽带隙钙钛矿室内光伏,通过多功能绝缘接触抑制漏电通道实现
暨南大学王有生/范建东/麦耀华等:超44%宽带隙钙钛矿室内光伏,通过多功能绝缘接触抑制漏电通道实现 海南大学张建、卢兴团队:几何结构与电子工程“双擎”驱动解码过氧化氢电合成催化剂设计关键法则
海南大学张建、卢兴团队:几何结构与电子工程“双擎”驱动解码过氧化氢电合成催化剂设计关键法则