Unlocking the Synergistic Promoter Role of Phosphorus in Evolving NiFe Phosphides for Enhanced Water Oxidation
Ningning Shi#, Mingcheng Gao#, M. Maneesha, C. S. Praveen*, Panpan Liu, Shengnan Yue, Wangjing Xie, Dechao Chen, Yu Tang, Yuanqing Wang*, Hua Fan & Xing Huang*
Nano-Micro Letters (2026)18: 400
https://doi.org/10.1007/s40820-026-02238-0
本文亮点
1. 磷诱导重构,协同稳定活性位:揭示了磷在NiFeP析氧催化剂动态演化中的关键促进作用。反应过程中,磷驱动NiFeP重构为高活性的NiFe(氧)氢氧化物,同时揭示磷化处理有效抑制Fe溶出,提高催化稳定性。
2. 缓冲氧化态,赋能高效析氧:PO₄³⁻与Fe协同构筑氧化还原缓冲体系,可避免Ni过度氧化,稳定活性位点;同时缩小电子带隙,提高导电性。重构后的催化剂展现出优异的碱性析氧性能,在10 mA cm⁻²下仅需225 mV的低过电位,并可在高达500 mA cm⁻²的电流密度下稳定运行超过100 h,表现出良好的高电流应用潜力。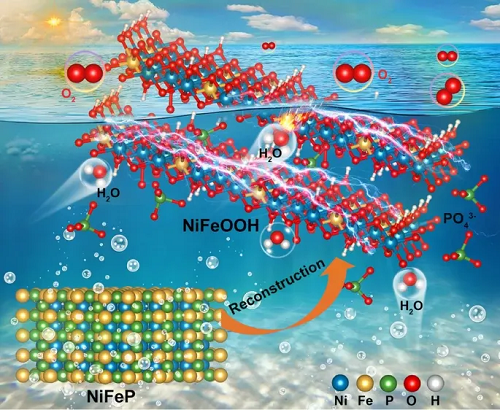
研究背景
氢能是支撑未来绿色能源体系的重要载体,而可再生电力驱动的电解水制氢被认为是获得绿色氢气的理想技术路径。然而,电解水过程中的阳极析氧反应动力学缓慢,严重限制了整体能量转换效率。目前高效析氧催化剂仍主要依赖Ru、Ir等贵金属材料,但其成本高、储量低,难以满足大规模应用需求。因此,发展廉价、高效且稳定的非贵金属析氧催化剂,是推动绿色制氢技术走向应用的关键。在众多候选材料中,NiFe基磷化物因具有良好的导电性、丰富的活性位点和优异的碱性析氧性能而备受关注。不过,近年来研究发现,金属磷化物在析氧反应中通常会发生原位结构重构,转化为真正发挥催化作用的金属(氧)氢氧化物。围绕这一过程,磷元素究竟只是促进重构的“牺牲组分”,还是能够进一步参与调控活性相的电子结构和稳定性,仍缺乏清晰认识。厘清磷在催化剂动态演化中的真实作用,对于设计新一代高效析氧催化剂具有重要意义。
内容简介
福州大学黄兴/上海大学王元庆/科钦科技大学C.S. Praveen等聚焦电解水制氢中阳极析氧反应动力学缓慢及贵金属催化剂成本高等关键问题,以NiFeP磷化物为模型体系,系统揭示了磷元素在催化剂电化学重构和析氧反应中的多重作用。研究发现,NiFeP在反应过程中会原位重构为高活性的NiFe(氧)氢氧化物相;同时,磷并非仅作为牺牲组分促进重构,其残留的PO₄³⁻物种能够与Fe协同调控Ni的电子结构、抑制Fe溶出,并通过氧化还原缓冲机制稳定活性位点。该工作阐明了“磷诱导重构—磷酸根协同调控−NiFe活性相稳定”的结构−性能关系,突破了以往将金属磷化物简单视为预催化剂或牺牲模板的传统认识,为理解阴离子在电催化动态重构中的作用提供了新视角,也为设计高效、稳定、低成本的非贵金属析氧催化剂提供了重要思路。
图文导读
I 模板诱导空心结构,铁磷协同调控电子态
如图1所示,研究团队通过牺牲模板转化和气相磷化策略,成功构筑了空心棱柱状NiFeP催化剂。该材料继承了NiFe前驱体的空心结构,具有粗糙表面和丰富暴露位点,有利于电解液渗透、活性位点暴露以及后续电化学重构。微观结构分析表明,NiFeP由晶态NixFeyP金属磷化物颗粒和富含Ni/Fe/O/P/K的非晶组分共同组成,形成晶态−非晶复合结构。XPS结果进一步证明,Fe的引入能够有效调控NiFeP的电子结构,增强Ni、Fe、P之间的电子耦合,为后续形成高活性NiFe(氧)氢氧化物相和提升析氧性能奠定了基础。
图1. 催化剂的合成与表征。
II 活化重构释放高效析氧活性,NiFeP实现低过电位与高电流稳定运行
从电化学性能来看,经电化学活化后的NiFeP-A催化剂展现出显著优于前驱体、氧化物、单金属磷化物以及商业RuO₂的OER活性,仅需225 mV过电位即可达到10 mA cm⁻²,且Tafel斜率低至 31 mV dec⁻¹,表明其具有快速的析氧反应动力学。
此外,NiFeP-A还表现出更低的电荷转移阻抗、更大的电化学活性面积以及更高的本征催化活性,在1.53 V vs. RHE下实现了23.63 mA cm⁻²的比活性、314.2 A g⁻¹的质量活性和0.14 s⁻¹的TOF,说明Fe引入与磷化处理协同提升了电子传输能力和活性位点利用效率。在稳定性方面,NiFeP-A可在10、100和500 mA cm⁻²等不同电流密度下连续稳定运行超过100 h,且经过10,000次循环后极化曲线几乎保持不变,体现出优异的耐久性。此外,在实际全解水装置中,以NiFeP-A作为阳极、商业Pt/C作为阴极构建的两电极体系仅需1.51 V即可达到10 mA cm⁻²,优于商业RuO₂||Pt/C基准体系。同时,该体系可连续运行100 h以上,显示出良好的应用潜力。上述结果表明,NiFeP经电化学活化后形成的真实活性结构能够同时实现高活性、快速动力学和长周期稳定性,为非贵金属催化剂实际应用提供了有力支撑。
图2. 催化性能评估。
III 原位重构显真身:NiFeP 演化为高活性NiFe(氧)氢氧化物
为揭示NiFeP在析氧反应中的真实活性结构,研究团队利用相同位置透射电镜(IL-TEM)跟踪其在阳极氧化过程中的形貌和组成演变。结果显示,初始空心棱柱状NiFeP在电化学氧化过程中逐步发生重构:随着反应时间延长,原有空心结构逐渐转变为片层状纳米结构,同时磷含量显著降低,而Ni/Fe比例基本保持稳定。这表明反应过程中磷发生部分溶出,而Ni、Fe金属骨架整体较为稳定。
相应的结构证据表明,经过电化学活化后,原始金属磷化物相基本消失,催化剂转化为低结晶度的NiFe基氢氧化物/(氧)氢氧化物纳米片。该重构结构富含缺陷,并保留少量均匀分布的磷物种。相关表征显示,残留磷可能以PO₄³⁻形式存在于片层结构中,不仅参与调控层间环境,也可能对稳定活性相发挥重要作用。由此可见,NiFeP并非在原始磷化物状态下直接发挥析氧催化作用,而是在阳极条件下原位重构为真正高活性的NiFe(氧)氢氧化物相。由电化学重构产生的低结晶、缺陷丰富和含磷调控的片层结构,为其优异OER活性提供了关键结构基础。
图3. 相同位置透射电镜(IL-TEM)研究。
IV 配位环境重塑,残留磷酸根稳定活性结构
为进一步确认NiFeP在析氧反应中的真实活性结构,研究团队利用XAFS、XPS和EELS等手段系统追踪了Ni的化学态和局域配位环境变化。结果显示,初始NiFeP中Ni主要与P配位,呈现典型金属磷化物特征;经过电化学活化后,Ni−P键基本消失,取而代之的是明显的Ni−O和Ni−(O)−M配位,其中M代表相邻的Ni或Fe原子。这表明NiFeP在反应过程中已由金属磷化物重构为NiFe基氧化物/氢氧化物/(氧)氢氧化物结构,与前述形貌演化结果一致。
谱学结果还揭示,活化后的NiFeP-A并非简单的NiFe氧化物,而是具有缺陷丰富、低结晶度和含磷调控特征的NiFe(氧)氢氧化物。XPS结果显示,反应后Ni−P和Fe−P物种消失,同时出现两类Ni2+物种:一类与晶格氧配位,另一类则可能与残留磷酸根等含氧阴离子配位。该结果说明,磷在反应中并未完全从催化剂中流失,而是部分转化为PO₄³⁻等含磷物种并保留在重构结构中,进一步调控Ni的局域电子环境。长时间稳定性测试后,催化剂仍保持片层形貌,残留磷物种依然均匀分布,整体化学态基本不变。该结果说明,电化学重构形成的NiFe(氧)氢氧化物活性相具有良好的结构稳定性,而残留PO₄³⁻可能在稳定活性结构、调控金属中心电子态和维持长期析氧性能方面发挥重要作用。
图4. 电子态和配位环境表征。
V 磷的双重角色:诱导重构并稳定活性相
为厘清磷在NiFeP析氧催化中的真实作用,研究团队对比了含磷和不含磷样品的电化学活化行为。结果表明,含磷的NiFeP和NixPy在反应过程中均发生明显结构重构,而不含磷的NiFeO则基本保持结构稳定,说明磷能够有效促进催化剂在阳极条件下发生结构演化,诱导形成更有利于析氧反应的活性相。进一步对比发现,未磷化的NiFe前驱体在活化过程中虽可转化为超薄Ni(氧)氢氧化物,但伴随严重Fe流失;而NiFeP活化后仍能较好保留Fe组分,说明磷化处理和热处理共同有助于稳定NiFe骨架。其中,磷的引入不仅促进了独特片层重构结构的形成,也提升了OER活性,证明磷化过程对于构筑高效催化剂至关重要。
更重要的是,NiFeP活化过程中虽然大部分磷会溶出,但少量残留磷酸根物种仍保留在催化剂层间结构中。同时,析出的P以PO₄³⁻形式在电解液中,可与催化剂表面直接接触。这些PO₄³⁻对催化稳定性和反应动力学可能发挥关键作用。通过向KOH电解液中引入PO₄³⁻,研究团队发现Ni基催化剂的OER活性明显提升,且存在最优磷酸根浓度;过量PO₄³⁻反而会削弱性能。这表明残留PO₄³⁻与NiFe活性相之间存在适度协同作用,可加速析氧反应过程并稳定催化活性。该结果进一步揭示,磷在NiFeP催化剂中具有双重作用:一方面诱导金属磷化物重构为高活性的NiFe(氧)氢氧化物,另一方面以PO₄³⁻形式参与调控活性相结构和界面反应环境,从而实现高活性与长稳定性的协同提升。
图5. 不同浓度PO₄³⁻性能比较。
VI “磷酸根不是旁观者,而是调控 NiFe 活性相的氧化还原缓冲因子”
理论计算从原子尺度解释了残留PO₄³⁻提升 NiFe(氧)氢氧化物析氧活性的内在机制。在析氧反应相关电位下,Fe掺杂能够降低Ni的平均氧化程度,而PO₄³⁻的引入则进一步稳定催化剂中的质子化状态,使Ni位点维持在更合适的电子结构区间。二者协同作用类似于“氧化还原缓冲器”,可避免Ni在反应过程中发生过度氧化,从而维持更稳定、更高效活性环境。
电子结构计算显示,PO₄³⁻和Fe能协同缩小NiOOH的带隙,增强电子离域和电荷传输能力,这与实验中NiFeP-A表现出更低电荷转移阻抗的结果相一致。同时,自由能计算表明,PO₄³⁻可调节*OH、*O和*OOH等关键析氧中间体的吸附强度,使反应能垒分布更加合理;当Fe与PO₄³⁻同时存在时,理论过电位由原始NiOOH的0.90 V显著降低至0.52 V,说明二者并非简单叠加,而是形成了强协同效应。
由理论结果可以得出,残留磷酸根不仅参与调控Ni的氧化还原状态,还能优化电子结构、促进电荷传输,并稳定关键OER中间体。由此,PO₄³⁻与Fe共同赋予重构后的NiFe(氧)氢氧化物更优的反应路径和更低的能量屏障,为实验中观察到的高析氧活性和长周期稳定性提供了清晰的理论依据。
图6. 理论计算。
VII 总结
本研究通过气相磷化策略构筑了空心棱柱状NiFeP催化剂,并揭示了其在碱性析氧反应中的动态重构过程。结果表明,NiFeP并非以原始磷化物结构直接发挥作用,而是在反应过程中原位转化为超薄NiFe(氧)氢氧化物纳米片。研究团队结合相同位置透射电镜、非原位 TEM、XAFS、XPS 和理论计算等手段,直接捕捉并阐明这一结构演化及其性能来源。
重构后的催化剂展现出优异的析氧活性和稳定性,在10 mA cm⁻²下仅需225 mV的低过电位,并可在高电流密度下稳定运行超过100 h。更关键的是,磷在该过程中发挥了多重作用:不仅促进活性 NiFe(氧)氢氧化物相的形成,还能抑制Fe溶出,并以残留PO₄³⁻物种的形式与Fe协同调控Ni的电子结构。理论计算揭示,PO₄³⁻与Fe可共同充当氧化还原缓冲因子,避免Ni过度氧化、增强电子传输,并稳定关键OER中间体,从而降低反应能垒。
该工作突破了将金属磷化物简单视为“预催化剂”或“牺牲模板”的传统认识,揭示了磷驱动结构重构和磷酸根协同稳定活性相的深层机制,为设计低成本、高性能、长寿命的非贵金属析氧催化剂提供了新的思路。
作者简介



关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc.),包括微纳米材料与结构的合成、表征、性能及其在能源、催化、环境、传感、人工智能、电磁波吸收与屏蔽、健康监测、生物医药等领域的应用研究及高水平综述。期刊已被SCI、EI、PubMed、SCOPUS等数据库收录,2025 JCR IF=38.5,学科排名Q1区前1.5%。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
期刊网址: https://springer.com/40820
投稿网址:https://mc03.manuscriptcentral.com/nmlett
E-mail: editorial_office@nmlett.org
Tel: 86-21-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 福州大学黄兴/上海大学王元庆/科钦科技大学C.S. Praveen等:揭示磷在NiFe磷化物结构演化与增强水氧化中的协同促进机制
 北理工胡斌/蔡然等:受猪笼草启发的智能“呼吸”电子皮肤
北理工胡斌/蔡然等:受猪笼草启发的智能“呼吸”电子皮肤 西工大黄河源&西交大赵鑫等综述:聚合物基柔性无线传感器——从材料设计到智能健康监测系统的全景综述
西工大黄河源&西交大赵鑫等综述:聚合物基柔性无线传感器——从材料设计到智能健康监测系统的全景综述 燕山大学杨成武/张新宇&泰国朱拉隆功大学秦家千等:自分离双相电解液打造电子富集界面,实现超长寿命水系锌离子电池
燕山大学杨成武/张新宇&泰国朱拉隆功大学秦家千等:自分离双相电解液打造电子富集界面,实现超长寿命水系锌离子电池 浙江大学于东旭、张雪艳、王利光:钠离子电池NASICON正极的结构、电化学与稳定性
浙江大学于东旭、张雪艳、王利光:钠离子电池NASICON正极的结构、电化学与稳定性