研究背景
水系锌离子/金属电池(Zinc ion/metal battery)因其水系电解液的高安全性以及锌电极的高经济性而备受关注。但其锌负极的不稳定性一直是阻碍其进一步商业化的重要问题。破坏其负极稳定性的主要原因是枝晶生长和各种副反应。其中枝晶生长源于电极表面微观缺陷导致的电场及锌离子分布的不均匀。而副反应则源于锌金属与电解液中水及盐分的相互作用。这些问题会导致活性物质的流失,电极表面的钝化,甚至长期的不均匀沉积会最终导致隔膜被刺穿。因此大家都在通过改善电解液,隔膜,电极表面等电池内一系列重要组分来诱导锌离子均匀沉积的同时抑制副反应的发生。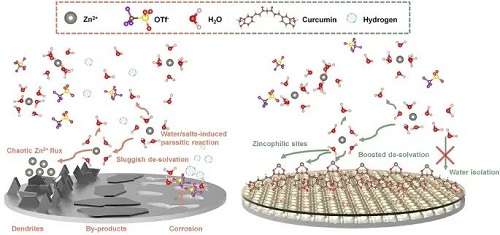
Creation of an Artificial Layer for Boosting Zn2⁺ Mass Transfer and Anode Stability in Aqueous Zinc Metal Batteries
Mingcong Tang, Qun Liu, Gang Liu, Xiaohong Zou, Kouer Zhang, Zhenlu Yu, Biao Zhang*, Liang An*
Nano-Micro Letters (2026)18: 121
https://doi.org/10.1007/s40820-025-01973-0
本文亮点
1. 加强锚点,有序沉积:β-二酮结构上存在一对对称的羰基,其较强的极性展现出了与锌离子较强的结合能力,通过将此结构的姜黄素与PVDF 粘结剂进行融合并均匀涂抹到锌箔表面,锌离子可以精准配位在两个羰基以上,使得双电层中锌离子的分布更加充裕且均匀。
2. 动态界面,加速传导:β-二酮结构具有独特的酮烯醇互变异构性,姜黄素分子会同时呈现出有质子配位的烯醇构型和无质子配位的酮构型。这两种构成的比例会因为周围环境的pH值而进行自发的动态调控,因此在电极表面就可以对Zn2⁺进行动态的传输。
内容简介
针对水系锌离子电池中负极锌枝晶生长和各种副反应导致的稳定性不理想,以及界面锌离子反应速率迟缓的问题,香港理工大学安亮教授团队和张标教授团队通过在负极表面使用超薄有机涂层修饰的方法,有效隔离锌金属和作为主要副反应来源的水,抑制了副反应的发生。因此也避免了锌电极表面因为腐蚀产物的产生而发生的钝化问题。同时通过姜黄素分子(CUR)上具有较高极性的双羰基,加强了对电解液界面的Zn2⁺的结合能力,使得Zn2⁺在双电层中的分布更加充沛且均匀。实现了负极表面的均匀沉积。同时β-二酮结构具有的独特的酮烯醇互变异构性会在有阳离子配位和脱配位的两种构型中进行动态调控,加速了界面Zn2⁺的传导。最终在实现了高负极稳定性的同时,有效降低了界面的脱溶剂化能垒以及活化能垒。
图文导读
I 动态界面调控:β-二酮结构加速锌离子的有序吸脱附
如图1所示,姜黄素分子(CUR)构成的涂层在锌负极表面,通过其结构上具有强极性的对称双羰基构建了丰富的Zn2⁺锚点用以实现对Zn2⁺的配位。同时其疏水的特性,有效屏蔽了电解液的中的水,避免了因为界面水电解而导致的析氢析氧。同时也避免了因为锌金属与电解液中的水和盐反应而产生钝化产物的问题。这种亲锌疏水性也引导了锌离子在界面反应的前的脱溶剂化过程中水分子的脱离,降低里整体的脱溶剂化能垒。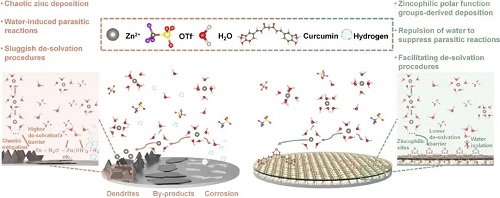
图1. 水系锌离子电池中负极的主要问题和本工作中提出的姜黄素涂层调控机制。
II 超薄简易屏障:CUR图层的构筑与其特性
本研究提出使用在水溶液中溶解度极低但在有机溶剂中溶解度极高的姜黄素(CUR)作为界面修饰材料。通过在NMP中溶解CUR和少量作为粘结剂的PVDF来制备的溶液,可以在涂层非常薄的情况下在表面施加丰富的活性物质(图2)。并且这种溶液态的前驱体可以确保最终制备出的表面涂层均匀且不容易脱落。最终只需要2 μm厚度的涂层就可以实现对锌负极的有效保护。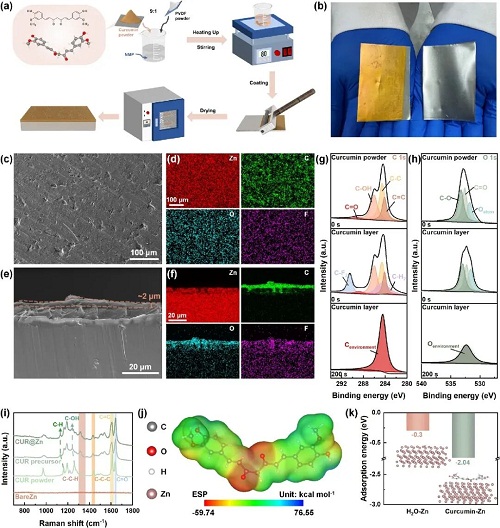
图2. CUR图层的制备过程和特性探索。
III 亲锌疏水+动态传输:抑制副反应的同时加速锌离子传导
为系统揭示姜黄素在锌电解剥离/沉积过程中的界面调控作用,作者通过电化学测试与理论计算对其界面机理进行了深入研究。首先,通过交流伏安测试考察姜黄素对锌表面界面环境的影响。与裸锌相比,CUR@Zn电极的界面电容明显降低,零电荷电位(PZC)由0.28 V移至0.24 V(图3a),表明姜黄素在锌表面发生了强烈吸附。并在随后评估了姜黄素对Zn2⁺迁移动力学的调控作用。通过对称电池(BareZn与CUR@Zn)的电荷转移电阻(Rct)测试,并结合温度与-ln(Rct)的线性关系,计算得到 CUR@Zn 电池的表观活化能仅为 14.56 kJ mol⁻1,远低于裸锌电极的38.57 kJ mol⁻1(图3c),说明界面 Zn2⁺传输显著加速。同时,锌离子迁移数从0.383提升至 0.435,进一步证明离子传导得到增强。
基于密度泛函理论(DFT)计算,进一步分析了Zn2⁺在不同表面上的吸附行为。在Zn(101)晶面上,Zn2⁺的吸附能Ea由裸锌的-1.01 eV 降至CUR@Zn的 -2.40 eV(图3e、3f),表明姜黄素增强了Zn2⁺在电极表面的吸附,有利于Zn2⁺从电解液向锌表面的迁移。Zn(002)和Zn(100)晶面上亦呈现类似趋势:吸附能分别由-0.5 降至-2.49 eV、由-2.59 降至-4.85 eV。由此可见,姜黄素能够在锌箔的主要晶面上普遍强化Zn2⁺吸附,促进致密、均匀的沉积结构,从而抑制枝晶生长。
为厘清实际配位构型,作者计算了Zn2⁺与姜黄素不同官能团的结合能,并与水和PVDF进行了对比。结合静电势(ESP)分布结果,选取C=O和-OH 作为潜在配位位点。计算显示,Zn2⁺-C=O的结合能为-0.93 eV,强于Zn2⁺-H₂O的-0.6 eV;Zn2⁺-OH的结合能为-0.52 eV。说明Zn2⁺更倾向与姜黄素配位,且优先与C=O基团结合。作者进一步计算了姜黄素与Zn2⁺配位前后体系的ESP变化。结果显示,配位后整个结构的 ESP 整体显著变负,说明Zn2⁺周围的静电排斥明显减弱,有利于离子传输。基于上述优势,姜黄素层有望加速Zn2⁺的脱溶剂化过程。图3j表明,在裸锌电极上,仅前两个水分子的剥离过程在热力学上较为“自发”,而之后每一步都需要较高的额外能量来驱逐更多水分子。相较之下,当[Zn(H₂O)₆]2⁺接近姜黄素时,可在无外加能量的情况下自发排出三个水分子;同时,实现完全脱溶剂化所需的总能量在CUR@Zn上显著降低,表明姜黄素有效促进了脱溶剂化过程。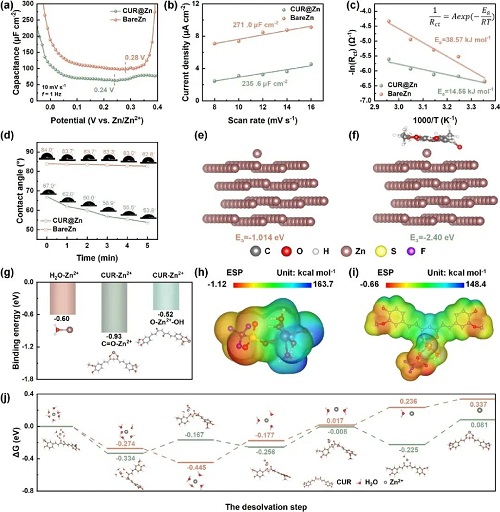
图3. CUR分子的量子化学性质。
IV 有效抑制副反应并实现均匀沉积
姜黄素涂层兼具本征化学惰性,因此作者首先通过线性扫描伏安(LSV)测试析氢反应(HER)与析氧反应(OER)的起始电位,并标定腐蚀电流和腐蚀电位,最终确定了更宽的电化学窗口以及大幅下降的腐蚀电流。综合来看,CUR@Zn既不易发生腐蚀,其腐蚀过程也更缓慢。光学显微与SEM图像也得到了相同结论。裸锌电极表面出现大面积暗灰色覆盖层;放大后可见大量片状腐蚀产物,平均尺寸约50μm。相比之下,CUR@Zn在光学与SEM图中均未见类似沉积,表面仍然光亮平整,与未处理锌箔几乎无异。
综上,姜黄素涂层通过提高界面化学惰性并选择性地与Zn2⁺相互作用,成功抑制了界面水诱导的电化学与化学副反应,同时通过“隔离效应”有效避免电解液对金属锌的化学侵蚀,使锌电极在水系环境中保持稳定、平整且免受腐蚀。
得益于姜黄素分子上的亲锌官能团所带来的增强Zn2⁺传输与均匀离子通量,锌在该界面上的沉积行为得到显著优化。形貌表征结果与理论预测高度一致。在首圈沉积时,CUR@Zn表面几乎被直径小于0.5μm的晶核均匀覆盖,即便生长一段时间后,沉积锌的特征尺寸也仅约5μm。相比之下,裸锌表面则可见大尺寸团聚体与明显的钝化区域,最大颗粒尺寸超过50μm,若继续累积便会演化为宏观枝晶。
为更直观展示姜黄素对离子分布及沉积行为的影响,作者利用COMSOL Multiphysics,对离子分布和沉积行为进行模拟。结果表明姜黄素层可将Zn2⁺ 的浓度分布从各向异性(集中在尖端)转变为近似各向同性。这样一来,Zn2⁺不再偏向积聚在局部突起位置、触发“尖端效应”,而是更均匀地供应于整体电极表面,使所有活性位点都有相对“公平”的沉积机会。由此,在初始阶段即可避免不均匀成核,并在长时间沉积中有效抑制枝晶的生成。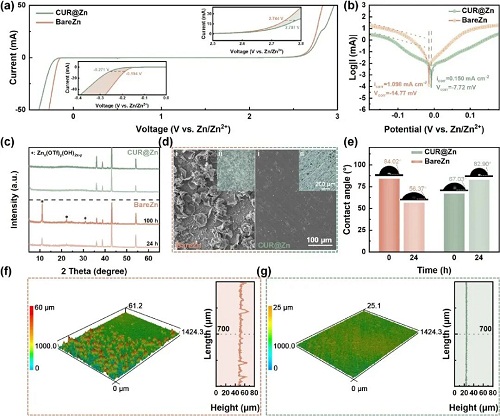
图4. CUR涂层对腐蚀的抑制。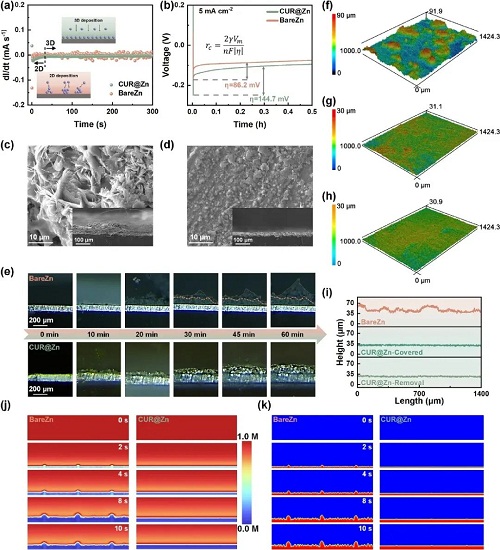
图5. CUR涂层对沉积行为的改善。
V 总结
本研究利用可降解的姜黄素构筑了一种高效的人造保护层,用于稳定锌负极。姜黄素与PVDF精细混合后可形成均匀的液态前驱体,通过简单涂布即可在锌表面构建致密且附着力优异的保护层。该有机骨架对Zn(OTf)₂电解液具有良好润湿性,而姜黄素分子上的极性官能团则充当Zn2⁺的锚定位点,从而显著提升电荷载流子的传输效率。
在仅约2 μm的厚度下,这个人造界面层即可有效屏蔽水诱导的副反应,并均匀调控Zn2⁺的通量,使锌的沉积更加平整致密。得益于此,CUR@Zn对称电池在 1 mA cm⁻2条件下可稳定运行超过2000小时;库仑效率在循环600圈后仍可达到99.15%,并实现60%的高放电深度,显著提高了锌的利用效率。在Zn||NVO全电池中,采用 CUR@Zn 负极同样表现出优异的循环寿命,在3000圈后仍能保持超过86.5%的容量。
综上,本工作通过简便的刮涂工艺,利用环境友好且可规模化的姜黄素构建了一种高效人造界面层,显著提升了锌负极的稳定性与可逆性,为Zn(OTf)₂基水系锌电池迈向商业化提供了一条切实可行的工程化路线。
作者简介


关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2024 JCR IF=36.3,学科排名Q1区前2%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 香港理工大学安亮/张标等:有机涂层β-二酮结构修饰锌负极促进水系锌电中动态Zn²⁺传输