研究背景
随着全球对可再生能源需求的增加,风能和太阳能等能源的间歇性和不稳定性带来了重大挑战。作为满足电网规模储能要求的首选,钠离子电池(SIBs)逐渐受到关注。在负极一侧,通常认为碳基材料具有较高理论容量和稳定循环性能。但不同碳结构的钠储存行为差异很大,例如Na在石墨中的插层表现一般,而容易嵌入到具有孔隙结构和扩大的层间距的硬碳(HC)中。HC的充放电曲线可分为斜坡区(>0.1 V)和平台区(<0.1 V),其中导致平台区产生的储钠机制一直是争论不断的中心话题。随着更广泛研究的进行,平台区主流观点已从层间插层转向闭孔填充。构建充足且合适的闭孔可以显著提高首次库伦效率(ICE)和平台容量。然而,对于闭孔结构和储存机制的理解分析仍然不够深入,进一步使得开孔高温下转为闭孔的过程缺乏系统解释。因此,迫切需要建立起统一的理论框架以解决争论难题和指导改性方法研究,推动具有高ICE和卓越能量密度的SIBs进一步发展。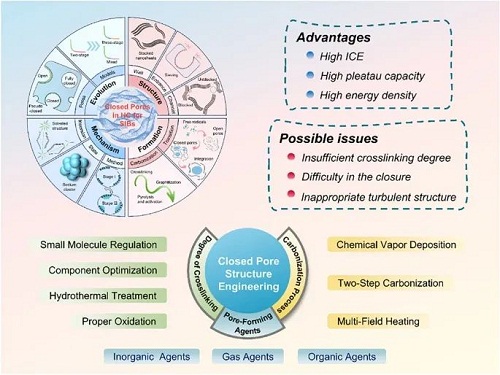
Comprehensive Understanding of Closed Pores in Hard Carbon Anode for High-Energy Sodium-Ion Batteries
Siyang Gan, Yujie Huang, Ningyun Hong, Yinghao Zhang, Bo Xiong, Zhi Zheng, Zidong He, Shengrui Gao, Wentao Deng, Guoqiang Zou, Hongshuai Hou*, Xiaobo Ji
Nano-Micro Letters (2025)17: 325
https://doi.org/10.1007/s40820-025-01833-x
本文亮点
1. 综述了钠离子电池硬碳负极内闭孔结构的最新进展,建立了活性位点统一视角下的概念框架和起源机制。
2. 系统探讨了闭孔特性对钠储存行为的影响,并提出了孔隙结构定向调控的设计原则。
3. 强调了未来可行的研究方向,将分子水平设计以及动力学/热力学混合分析运用于先进的修饰策略以实现性能优化。
内容简介
硬碳(HC)因其高成本效益和出色的整体性能而被认为是最有前途的钠离子电池(SIBs)负极材料。然而,HC的无定形的复杂微观结构对阐明结构-性能关系提出了重大挑战,这导致了对闭孔内在特性的持续误解。不合理的闭孔构造方法必然会导致平台容量下降,严重制约了HC在高能量密度场景下的实际应用。中南大学侯红帅&纪效波等人系统阐述了闭孔的概念框架和起源机制,为它们的结构特征和形成途径提供了重要见解。随后,通过将晶格参数与缺陷构型相关联,严格建立了控制脱溶剂化动力学和钠储存行为的结构-性能关系。此外,归纳了闭孔结构工程的开创性进展,以建立合理调节HC中闭孔的基本设计原则。研究者指出,采用分子水平的视角,结合协同动力学/热力学方法,对于理解和控制从开孔到闭孔的转化过程至关重要。
图文导读
I 闭孔的概念和特性
在设计和生成优秀的闭孔之前,全面理解闭孔的概念和特性是至关重要的。尤其是孔结构的研究历史、不同孔结构的定义、闭孔的参数、钠存储机制和形成过程(图1)。目前,HC的理想微观结构广泛被认同如下:在局部尺度上,由略微扭曲的石墨烯纳米片组成的条纹状伪石墨微晶以几层堆叠(通常为2-4层)形成,由于存在缺陷,展现出短程有序。在更大尺度上,这些微晶作为基本的结构单元不规则分布,形成的湍流结构构建出众多开放和闭合纳米孔。HC在X射线衍射(XRD)图谱中通常表现出约24°和43°的两个特征宽衍射峰,可进一步计算出石墨微晶的平均层间距(d₀₀₂)、平均宽度(La)、平均厚度(Lc)和堆叠数(n)。晶体结构的特性决定了这些孔的具体特征,包括孔壁的曲率、孔入口、孔通道和孔径大小,它们是温度依赖的。当采取较高的碳化温度时,理想的闭孔形成过程包括三个阶段:开孔的产生(热解释放自由基)、弯曲孔壁的发展(缺陷修复)和孔结构的演变(孔隙合并和收缩)。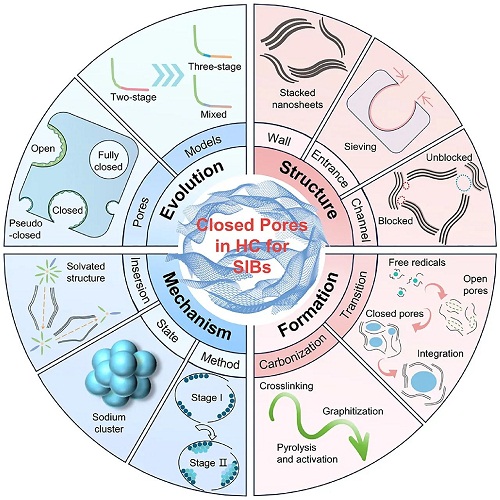
图1. SIBs中硬碳闭孔的演变、结构、机制和形成过程的示意图。
根据不同的孔隙参数,本文重申了“开孔”和“闭孔”的概念并提出“准闭孔”和“完全闭孔”进行补充(见图2),具体来说:i) 在低碳化温度下制备的HC材料几乎完全为开孔,纳米片之间的距离相对较大,允许与外部气体接触,并完全暴露于外部电解质中。放孔仅对斜率容量有贡献,导致非常低的ICE和平台容量。ii) 如果热处理温度适中,所得HC材料的纳米片之间的间隙部分收缩,形成准闭孔(包括超微孔)。准闭孔的弯曲纳米片之间保持一定的空间,允许气体检测,部分反映了闭孔的整体大小和形状。一些电解质会被阻塞,对应于一些斜率容量转化为平台容量。iii) 在较高的制备温度下,闭孔通过石墨微晶的紧密互锁形成,只留下狭窄的间隙。在这个阶段,气体进入被禁止(通常指N₂),溶剂分子也受到极大限制。这些理想的闭孔能够显著延长平台容量。iv) 当碳化温度超过临界阈值(取决于前驱体)时,一部分闭孔转变为完全闭孔,甚至无法接触到电解质,平台容量反而下降。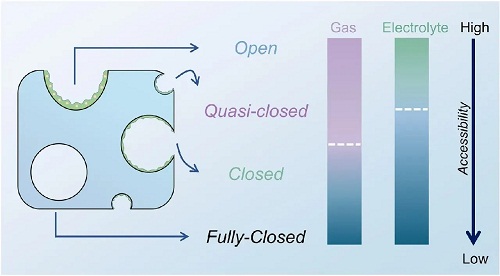
图2. 基于流体可及性的孔隙分类示意图。白色虚线代表边界,紫色和绿色分别表示气体和电解质的可接近性,蓝色表示不可接近。
ICE的增加源于闭孔间隙的空间限制,这种限制阻碍了在首次循环中(图3a)纳米孔内部壁面上不良固体电解质界面(SEI)的形成。然而,单纯的SEI不能解释所有实验观察结果,因为其固有的逻辑框架有限。例如,具有相同SBET值的样品在ICE上显示出显著差异。闭孔中的关键活性位点与放孔隙中的位点不同,因为它们必须首先穿过狭窄的孔入口。因此,越来越多的研究人员承认溶剂化过程的关键作用,并努力阐明电解液的溶剂化结构。溶剂化过程描述了钠离子在有机溶剂中溶解以形成溶剂化结构,随后在电解液-电极界面发生迁移和脱溶剂化。在溶剂化结构中,钠离子被两个溶剂化壳围绕,溶剂分子和阴离子通过静电力和配位键与钠离子相互作用(图3b)。以超微孔为例,这些微孔可以作为分子筛或离子筛,而不会妥协离子扩散能力。类似地,其他较大的闭孔也能由于孔口的存在而实现类似的筛分效应。不同碳化温度下制备的HC电极横截面上各种元素的信号强度显著不同(图3c)。在低碳化温度下,碳和钠信号的分布较为相似,归因于溶剂分子的渗透。相比之下,高碳化温度制备的HC中钠信号主要出现在低碳信号强度的区域,表明钠的损失主要发生在颗粒的外表面。除孔口外,溶剂化结构的大小是影响钠插入过程的关键因素。具体而言,较大尺寸的溶剂分子会在闭孔入口的几何约束下无法进入,有助于理想SEI的形成。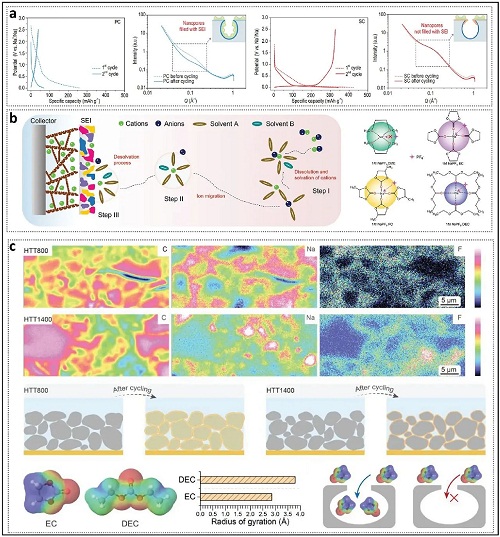
图3. 溶剂化过程和SEI的形成。a 展示了不同孔结构的HC在前两次充放电周期和五个完整循环后的SAXS图谱。b 说明了配位结构和溶剂化过程对电化学性能的影响。c 展示了HTT800和HTT1400的EPMA表征,以及通过理论计算确定尺寸的溶剂化结构在通过闭孔时的性能。
II 闭孔结构工程
形成闭孔的热解和石墨化过程与自由基的反应性和交联系统的稳定性密切相关。因此,碳化过程可以视为化学动力学和热力学的综合结果。由于不同前驱体类型的高度变异性和固有限制,传统的直接碳化方法常常引入几个关键问题:
i) 交联度不足,使碳材料容易发生剧烈重排,形成孔缺乏的软碳;
ii) 孔径过大或浓度过高的开孔,妨碍它们转化为闭孔。这些问题可归因于热力学与动力学之间不可逆的相互作用;
iii) 激活不足、石墨化过度以及孔径过大所导致的不适当湍流结构,阻碍了闭孔中钠离子存储效率。
为了展现HC的优秀电化学性能,特别是在低电压平台容量上的表现,许多闭孔结构工程策略被提出(图4)。这些策略可分为两种主要方法,具体取决于改性是在前驱体阶段还是在碳化过程中进行的。晶体参数调整(d₀₀₂、La、Lc)、分子结构控制和表面化学优化等多种因素需要被全面的考虑在内,以建立一个良好调控的系统指导闭孔结构的合理设计。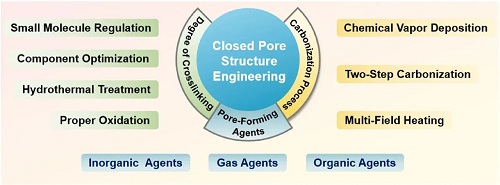
图4. 闭孔结构工程的分类,包括交联度调控、孔形成剂添加和碳化过程控制。
前驱体改性是在退火前对前驱体进行结构调整,例如交联或孔形成策略。交联是指多个线性聚合物链自组装并发生聚缩合反应,通过侧链上官能团的连接形成网络聚合物。在没有掺杂异原子(如氮、磷)的情况下,前驱体中的交联主要通过羰基和羟基位点来促进。在热解过程中,交联度的调整影响着不同化学键的断裂能力和骨架结构的整体刚性。如果交联度不足,在热解过程中自由基会剧烈释放,导致过多的孔产生,并可能使碳材料熔化并失去原有形态。而高度交联的样品在热解过程中经历了温和且均匀的过程,促进了低温微孔和高温闭孔的形成。调节交联的方式通常有预氧化、水热处理、组分优化和小分子调节,适用于不同的前驱体。此外,一些前驱体材料中的孔形成来源仍然有限,无法显著增强平台容量。因此,引入孔形成剂(或活化剂)被广泛认为是提高闭孔工程中孔隙率和孔体积的最直接高效的策略。成孔剂按种类可分类为气体成孔剂、无机成孔剂和有机成孔剂。值得注意的是,孔形成剂不仅有助于孔体积的增加,还能增强整体无序度。过程控制改性是指调节热解孔形成速率,并在前驱体碳化过程中优化石墨化程度,最终得到高质量HC。含有高浓度开孔的前驱体材料(例如多孔碳)通常需要经过非常高的煅烧温度,这使得它们不适合实际应用。此外,如果加热速率和持续时间没有进行调整,碳化过程主要由石墨化反应的热力学控制。较低的温度可能导致部分孔口未完全闭合,而较高的温度则存在过度缺陷修复和闭孔形成的风险,极大地限制了微观结构的可控性。为了充分发挥硬碳的潜力以实现其结构特性和电化学性能的全面优化,必须采用多种碳化技术相结合的方法。
III 总结
近年来,闭孔结构的发现极大地增强HC的研究活力和应用潜力。机遇与挑战并存,本文总结了该领域的最新研究进展,首先从机理的角度详细解释了闭孔结构的关键作用。通过对钠储存机制的深入分析,系统地揭示了闭孔的关键特性。其次,综述了闭孔结构的各种设计策略,为未来的结构调控提供了有价值的指导。总之,由于其强大的低电位充放电平台和优异的脱溶解能力,具有先进闭孔的HC在钠离子电池中展现了巨大潜力。虽然本文为闭孔结构工程提供了较全面的技术体系,然而决定闭孔实际性能的因素错综复杂,一定程度上阻碍了HC的商业化能力。未来仍然需要在分子级优化前驱体配置、探究孔成形剂的普适规律、结合动力学与热力学分析、开展缺陷工程深入闭孔填充机制和推进电解液工程加深对溶剂化过程的理解等领域不断探索。
作者简介

关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2024 JCR IF=36.3,学科排名Q1区前2%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 中南大学侯红帅/纪效波等综述:高能钠离子电池硬碳负极中闭孔的全面理解
 中科院苏州纳米所吴晓东&天目湖先进储能院王志诚团队:稀释剂—阴离子解耦离子液体电解液,实现高压高安全锂金属电池
中科院苏州纳米所吴晓东&天目湖先进储能院王志诚团队:稀释剂—阴离子解耦离子液体电解液,实现高压高安全锂金属电池 东北大学青勇权: 超疏水可穿戴应变传感器:从设计到高稳定范式
东北大学青勇权: 超疏水可穿戴应变传感器:从设计到高稳定范式 High Polarity Doping of CoFe Layered Hydroxides: Bifunctional and Corrosion Resistant Anion Exchange Membrane Seawater Electrolyzers Anandhan Ayyappan Saj, Sampath Prabhakaran, Mohsin Rasool, Kousik Bhunia, Dongho Lee, Hyunseok Ko, Tukaram D. Dongale, Muthukumar Perumalsamy, Arul Saravanan Raaju Sundhar, Do Hwan Kim & Sang Jae Kim Nano-Micro Letters (2026)18: 393 https://doi.org/10.1007/s40820-026-02230-8 本文亮点 1. 催化机理优化:本研究采用氧化镁纳米颗粒辅助法制备钴铁层状金属氢氧化物(CoFe LMH);氟掺杂可选择性调控铁位点并保持钴位点结构完整,有效改性钴铁层状氢氧化物的电子结构,使铁原子形成高自旋构型,进而提升催化活性。 2. 抗腐蚀能力突出:本研究通过氟掺杂改性样品 F-CoFe LMH-8 为双功能催化剂,在海水环境中兼具优异的亲水性、亲氧性,同时具备显著的疏氯特性,可有效排斥氯离子。 3. 长周期运行稳定:本研究以 F-CoFe LMH-8 同时作为阴、阳极组装的阴离子交换膜电解装置,在海水电解体系中,施加 2.3 V 电压即可实现1 A⋅cm⁻²的电流密度;装置连续运行 500 小时,电压衰减率仅为0.15 μV⋅h⁻¹,展现出极佳的运行稳定性。 微信图片_2026-06-23_091114_955.png 研究背景 氢能被视作清洁能源核心载体,电解水制氢是实现 “氢经济” 的主流技术,传统电解水依赖大量淡水资源,大规模应用受限。而海水储量丰富、取用便捷,直接海水电解成为替代淡水制氢、降低成本的理想路线,但面临两大核心难题:一是海水中大量氯离子会引发电极腐蚀、发生竞争性析氯副反应,严重破坏催化剂结构、降低产氢效率;二是析氢反应(HER)与析氧反应(OER)动力学缓慢,现有非贵金属催化剂活性不足、稳定性差。 目前主流层状双氢氧化物(LDH)类电极材料,在碱性海水体系中易发生结构重构,氯离子极易侵蚀活性位点;各类阴离子插层、表面改性等手段虽有改善,但普遍存在工作电压高、稳定时长短、难以工业化的问题。而贵金属催化剂活性优异,但价格昂贵、储量稀缺,无法满足规模化海水电解需求,因此开发低成本、高活性、耐氯腐蚀的非贵金属双功能电解催化剂具有重要现实意义。 内容简介 海水电解制绿氢是一项极具发展前景的技术,但OER动力学迟缓、氯离子腐蚀等问题严重制约了其实际应用。全北国立大学Do Hwan Kim等制备了一种新型氟掺杂钴铁层状金属氢氧化物催化剂(F-CoFe LMH-8),该材料是性能优异的双功能电催化剂。在电流密度为10 mA⋅cm⁻²条件下,其HER、OER的过电位分别仅为 81.23 mV 和 265.5 mV。理论计算与实验结果表明:氟掺杂可调控材料电子结构,使铁活性位点转变为高自旋构型,优化反应中间体吸附能,同时赋予材料疏氯特性,有效抵御氯离子带来的腐蚀。将 F-CoFe LMH-8 同时作为阴、阳极双功能催化剂,组装得到阴离子交换膜水电解槽(AEMWE),在连续制氢过程中表现出优异性能:在 1 mol/L KOH溶液中,电流密度可达1.2 A⋅cm⁻²;在 1 mol/L KOH+0.5 mol/L NaCl混合溶液中,电流密度为1.02 A⋅cm⁻²;在海水基 1 mol/L KOH体系中,施加 2.3 V 电压即可实现1 A⋅cm⁻²的电流密度。此外,研究采用长短期记忆(LSTM)机器学习模型对 F-CoFe LMH-8 的稳定性进行预测。该研究思路为定向设计适用于海水电解阴离子交换膜电解槽(AEMWE)、可规模化制氢的高稳定性、疏氯型高性能电催化剂,提供了一套完整的研究方案。 图文导读 I F-CoFe LMH的制备与表征 图1展示了氟掺杂钴铁层状氢氧化物的合成流程与物化性质。本研究采用氧化镁纳米颗粒辅助法制备钴铁层状金属氢氧化物(CoFe LMH)材料,高分辨透射电镜(TEM)观测结果显示(图1b-e),氟掺杂并未破坏材料原本的纳米片形貌:其中未掺杂样品 CoFe LMH-3 的晶面间距为 0.2521 nm,氟改性后的 F-CoFe LMH-8 晶面间距增至 0.2612 nm,直观证明掺杂后材料晶格发生膨胀。结合XRD、Raman光谱与傅里叶变换红外光谱(图1f-h)测试结果可知,氟元素成功掺入晶体晶格且未生成其他杂相,同时有效调控了材料内部金属 – 氧键、羟基等化学键与表面官能团结构。图1i结合电子顺磁共振结果进一步证实,F-CoFe LMH-8 的信号强度显著高于原始样品,说明氟掺杂成功诱导铁原子形成高自旋构型,这一结构特征是该催化剂能够展现优异电催化性能的核心原因。 1.png 图1 (a) 材料合成示意图;(b、c) CoFe LMH-3 在不同放大倍数下的高分辨透射电镜图;(d、e) F-CoFe LMH-8 在不同放大倍数下的高分辨透射电镜图(插图为对应的选区电子衍射图谱);CoFe LMH-3 与 F-CoFe LMH-8 的:(f) 粉末 XRD及对应晶面间距、(g) Raman光谱、(h) 傅里叶变换红外光谱、(i) 电子顺磁共振图谱。 图2通过多种表征手段,深入分析了氟掺杂前后钴铁层状氢氧化物的元素化学态、原子配位环境与电子结构特征。XPS谱图分别采集了Co、Fe、O、F四种元素的精细谱图(图2a-d),对比测试结果可见,氟掺杂后钴元素的特征峰位置、峰形均未发生明显偏移,证明钴位点的化学价态与局域电子环境基本保持不变;而铁元素的特征衍射峰出现显著位移,结合能发生规律性改变,直观反映出氟掺杂对铁原子电子云分布产生了明显调控。由此可见氟掺杂对钴元素的电子环境影响微弱,但会显著改变铁元素的结合能,证明氟原子可选择性作用于铁位点,同时完整保留钴活性中心的结构。同时图谱中出现了清晰的氟元素特征峰,也直接证实氟原子成功负载于材料表面并参与结构构建。 在此基础上,图2e-l通过X 射线吸收近边结构(XANES)与扩展 X 射线吸收精细结构(EXAFS)测试进一步印证了这一规律:钴的 K 边吸收谱与配位键长基本无变化,而铁的 K 边吸收峰向高能方向偏移,Fe-O 键作用明显增强,金属间键合被弱化。结合小波变换扩展 X 射线吸收精细结构图谱能够看出,氟掺杂精准调控了铁的局域配位环境,最终诱导铁形成高自旋状态,从原子层面揭示了材料催化性能提升的内在机理。 2.png 图2 CoFe LMH-3 与 F-CoFe LMH-8 的 XPS 精细谱:(a-d) Co、Fe、O、F;(e) 钴 K 边 X 射线吸收近边结构谱及 (f) 对应的扩展 X 射线吸收精细结构谱;(g) 铁 K 边 X 射线吸收近边结构谱及 (h) 对应的扩展 X 射线吸收精细结构谱;钴 K 边小波变换扩展 X 射线吸收精细结构谱:(i) CoFe LMH-3、(j) F-CoFe LMH-8;铁 K 边小波变换扩展 X 射线吸收精细结构谱:(k) CoFe LMH-3、(l) F-CoFe LMH-8。 II F-CoFe LMH的电化学性能 图3综合展示了系列催化剂的电化学性能、界面反应特征与理论吸附特性。图 3a、3b分别为线性扫描伏安曲线与塔菲尔斜率,结果表明氟掺杂改性后的 F-CoFe LMH-8 拥有更优异的反应动力学性能,电荷转移阻力更低,电催化反应速率远优于未掺杂的 CoFe LMH-3。图 3c奈奎斯特图同样印证了该结论,F-CoFe LMH-8 的圆弧半径明显更小,代表材料界面电荷传输效率更高、导电性能更佳。 图 3d、3e 与 3f、3g为扫描电化学显微镜测试结果,分别呈现了两种材料的基底电流与探针电流分布。F-CoFe LMH-8 的电流响应强度显著更高,说明其表面活性位点数量丰富,且反应活性分布均匀。图 3h是 F-CoFe LMH-8 在恒定电流密度−50 mA⋅cm −2下的电化学稳定性测试,全程电位波动极小,体现出出色的电化学耐久性能。 结合理论计算,图 3i、3j、3k分析了氢吸附中间体在钴、铁、氧位点的吸附能与吉布斯自由能:铁位点对氢中间体的吸附强度最为适中,既保障反应顺利推进,又不会因吸附过强占据活性位点,从理论层面解释了该材料析氢催化活性突出的内在原因。 3.png 图3 (a) 不同组分氟掺杂与未掺杂钴铁层状氢氧化物催化剂的线性扫描伏安曲线;(b) 对应的塔菲尔斜率;(c) CoFe LMH-3 与 F-CoFe LMH-8 的奈奎斯特图;CoFe LMH-3 的扫描电化学显微镜测试:(d) 基底电流分布、(e) 探针电流分布;F-CoFe LMH-8 的扫描电化学显微镜测试:(f) 基底电流分布、(g) 探针电流分布;(h) F-CoFe LMH-8 在恒定电流密度 −50 mA⋅cm⁻² 下的电化学稳定性测试;氟掺杂钴铁层状氢氧化物催化剂上氢吸附中间体在 (i) 钴位点、(j) 铁位点、(k) 氧位点的吸附能;钴、铁、氧位点上氢吸附中间体的吉布斯自由能。 图4围绕不同组分催化剂的析氧电化学性能、反应机理及电子结构展开分析。图 4a、4b为各样品的线性扫描伏安曲线与塔菲尔斜率,对比多款未掺杂、氟掺杂钴铁层状氢氧化物可以发现,F-CoFe LMH-8 的起始电位更低、塔菲尔斜率更小,代表其OER动力学最优,催化反应速率领先其余样品。图 4c的奈奎斯特图进一步佐证了上述结论,相较于 CoFe LMH-3,F-CoFe LMH-8 的阻抗圆弧半径更小,界面电荷转移阻力更低,电子传输能力更强。图 4d、4e和图 4f、4g为扫描电化学显微镜测试结果,F-CoFe LMH-8 的基底电流与探针电流信号更强,说明材料表面活性位点分布均匀且本征活性更高。 图 4h的稳定性测试结果显示,F-CoFe LMH-8 在持续工作过程中性能保持稳定,具备良好的长效服役能力。结合图 4iOER机理图与图 4j吉布斯自由能图,可明确材料的反应路径与最优活性位点。图 4k、4l的分态密度图谱则揭示了电子结构差异:氟掺杂有效调控了费米能级附近的电子态密度,提升了电子迁移能力,最终从电子层面阐明了 F-CoFe LMH-8 高效催化OER的本质。 4.png 图4 (a) 多种样品的线性扫描伏安曲线;(b) CLMH、CoFe LMH-1、CoFe LMH-2、CoFe LMH-3、CoFe LMH-4、FLMH-5、F-CLMH-6、F-CoFe LMH-6、F-CoFe LMH-7、F-CoFe LMH-8、F-CoFe LMH-9 及 F-FLMH-10 的塔菲尔斜率;(c) CoFe LMH-3 与 F-CoFe LMH-8 的奈奎斯特图;CoFe LMH-3 的扫描电化学显微镜测试:(d) 基底电流、(e) 探针电流;F-CoFe LMH-8 的扫描电化学显微镜测试:(f) 基底电流、(g) 探针电流;(h) F-CoFe LMH-8 的稳定性测试;(i) OER机理;(j) F-CoFe LMH OER的吉布斯自由能图(用于确定反应活性位点);(k) CoFe LMH-3、(l) F-CoFe LMH-8 不同配位位点的分态密度图。 III F-CoFe LMH的耐蚀性测试与机理 图5聚焦催化剂在海水体系下的耐腐蚀性、电化学行为与作用机制。图 5a测试了 F-CoFe LMH-8 在纯KOH溶液、模拟海水以及添加天然海水的电解液中的线性扫描伏安曲线,样品在三类体系中均保持稳定的电化学响应,初步体现出良好的海水环境适应性。图 5b为两种材料在模拟海水中的线性极化曲线,相比 CoFe LMH-3,F-CoFe LMH-8 的腐蚀电流更低,抗腐蚀能力大幅提升。图 5c氯离子吸附能图谱直观说明,F-CoFe LMH-8 对氯离子的吸附作用较弱,能够有效减少氯离子在材料表面附着,这也是其具备优异疏氯性能的关键。图 5d奈奎斯特图与图 5e弛豫时间分布分析结果显示,氟掺杂改性后材料的阻抗特征更优,界面结构在模拟海水中不易被破坏。图 5f、5g多电位相频特性曲线进一步反映出,F-CoFe LMH-8 在不同电位下电化学界面状态更稳定。 图 5h原位拉曼光谱证明,长期处于模拟海水环境中,材料的晶体结构与化学键未发生明显改变,结构稳定性优异;图 5i长效稳定性测试也验证了该结果。图 5j–5q为不同电位下的扫描电化学显微镜测试数据,在 0.3 V 和 0.35 V(vs Ag/AgCl)电位条件下,F-CoFe LMH-8 的基底电流与探针电流信号始终稳定且强度更高,表明其表面活性位点在海水体系中可正常发挥作用。最后,图 5r为离子作用机理示意图,清晰阐释了材料排斥氯离子、保障催化反应持续进行的微观原理。 5.png 图5 (a) F-CoFe LMH-8 在 1 mol/L KOH溶液、模拟海水(1 mol/L KOH+0.5 mol/L NaCl)以及天然海水配制的 1 mol/L KOH 溶液中的线性扫描伏安曲线;(b) CoFe LMH-3 与 F-CoFe LMH-8 在模拟海水中的线性极化曲线;(c) 氯离子吸附能图谱;(d) 奈奎斯特图;(e) CoFe LMH-3 与 F-CoFe LMH-8 的弛豫时间分布分析;(f) CoFe LMH-3、(g) F-CoFe LMH-8 的多电位相频特性分析;(h) F-CoFe LMH-8 在模拟海水中的原位拉曼测试结果;(i) 其电化学稳定性测试结果。在参比电极银 / 氯化银电位为 0.3 V 条件下,CoFe LMH-3 的扫描电化学显微镜测试:(j) 基底电流、(k) 探针电流;电位升至 0.35 V 时:(l) 基底电流、(m) 探针电流。在 0.3 V(vs Ag/AgCl)条件下,F-CoFe LMH-8 的扫描电化学显微镜测试:(n) 基底电流、(o) 探针电流;电位升至 0.35 V 时:(p) 基底电流、(q) 探针电流。(r) 离子在 F-CoFe LMH-8 材料表面的作用机理示意图。 IV F-CoFe LMH的稳定性测试 图6主要探究了催化剂在电解液及电解器件中的综合性能、长效稳定性与运行表现。图 6a、6b是 CoFe LMH-3 与 F-CoFe LMH-8 在模拟海水电解液(1 mol/L KOH+0.5 mol/L NaCl)中的线性扫描伏安曲线与多阶电流密度稳定性测试,对比可见 F-CoFe LMH-8 电化学活性更出色,在阶梯式变化的电流工况下性能也能平稳维持。图 6c进一步开展大电流稳定性测试,该样品在200 mA⋅cm⁻²、400 mA⋅cm⁻²高电流密度下持续工作,电位无明显波动,耐受大电流冲击的能力较强。 图 6d为阴离子交换膜水电解槽(AEMWE)装置示意图,直观展示了器件整体结构。图 6e、6f分别是搭载 F-CoFe LMH-8 双功能催化剂的电解槽,在三种不同电解液体系下的极化曲线与奈奎斯特图,结果表明该电解槽在常规碱液、模拟海水、天然海水体系中均拥有较低的极化电压与界面阻抗,环境适配性良好。 图 6g记录了电解槽的长期运行稳定性,器件可连续稳定工作。图 6h将电解槽电压衰减率与美国能源部(DOE)既定指标进行对比,本体系衰减速率远低于标准要求,长效运行优势显著。图 6i针对海水体系下的电解槽开展不同运行时长的弛豫时间分布分析,数据证明随着运行时间增加,器件界面结构与电化学特性未出现明显劣化。图 6j借助时序分析法对催化剂及电解器件的长期稳定性进行预测,进一步佐证了该材料在海水电解规模化应用中的潜力。 6.png 图 6 (a) CoFe LMH-3 与 F-CoFe LMH-8 催化剂在 1 mol/L KOH+0.5 mol/L NaCl体系中的线性扫描伏安曲线;(b) 多阶电流密度下的稳定性测试结果;(c) F-CoFe LMH-8 在上述电解液中,分别恒定电流密度为 200 mA⋅cm⁻²、400 mA⋅cm⁻² 时的稳定性测试;(d) 阴离子交换膜水电解槽(AEMWE)装置示意图;(e) 采用 F-CoFe LMH-8 双功能催化剂组装的电解槽,分别在 1 mol/L KOH、1 mol/L KOH+0.5 mol/L NaCl、1 mol/L KOH+天然海水体系下的极化曲线;(f) 对应的奈奎斯特图;(g) 该电解槽的长期运行稳定性;(h) 电压衰减率与美国能源部(DOE)技术指标对比;(i) F-CoFe LMH-8 基电解槽在海水体系中不同运行时长下的弛豫时间分布分析;(j) 借助时序分析法对催化剂稳定性进行的预测结果。 V 总结 综上所述,本研究对钴铁层状金属氢氧化物进行氟掺杂改性,成功制备出一种性能优异、稳定性强的双功能电催化剂,可应用于长效海水电解体系。氟掺杂可精准调控钴铁层状氢氧化物的铁配位位点,同时不会破坏用于OER的钴活性金属中心。所制备的 F-CoFe LMH-8 兼具出色的析氢与析氧催化性能:电流密度达到10 mA⋅cm⁻²时,析氢过电位为 81.23 mV,析氧过电位为 265.5 mV。氟掺杂能够强化铁位点的铁 – 氧键作用,促使双电层内氢氧根离子持续富集,有效抑制氯离子腐蚀,进而提升催化剂的催化活性与服役寿命。此外,采用 F-CoFe LMH-8 同时作为阴、阳极组装的阴离子交换膜海水电解槽,在 2.3 V 电压下可实现1 A⋅cm⁻²的电流密度;在0.125 A⋅cm⁻²恒定电流下连续运行 500 小时,电压衰减率仅为0.15 μV⋅h⁻¹,不仅达到美国能源部相关技术指标,也证明该体系具备工业化应用潜力。本研究为简易制备高性能、抗腐蚀海水电解催化剂提供了新思路,可推动阴离子交换膜海水电解槽的长效稳定运行。 作者简介 7.jpg图片 Do Hwan Kim 本文通讯作者 韩国全北国立大学 教授 ▍主要研究领域 电催化水分解、海水电解、阴离子交换膜电解槽技术以及非贵金属催化材料设计。 ▍主要研究成果 Do Hwan Kim教授任职于韩国全北国立大学,为科学教育学部教授、表面化学实验室负责人,主要研究方向涵盖电催化水分解、海水电解、阴离子交换膜电解槽技术以及非贵金属催化材料设计,在Adv. Funct. Mater.、Nano Energy 等知名期刊累计发表 SCI 论文 130 余篇,总引用量超 5000 次,h 指数约 40,研究成果多见于,在本研究中主要负责催化剂设计、电子结构调控与电解槽性能测试分析。 ▍Email:dhk201@jbnu.ac.kr 图片8.png Sang Jae Kim 本文通讯作者 韩国济州国立大学 教授 ▍主要研究领域 海水电解双功能催化剂、层状氢氧化物复合材料、电极界面调控及抗氯离子腐蚀技术。 ▍主要研究成果 Sang Jae Kim教授毕业于日本东北大学,曾赴剑桥大学、佐治亚理工学院开展访学研究,现任韩国济州国立大学教授、纳米材料与系统实验室负责人,长期致力于海水电解双功能催化剂、层状氢氧化物复合材料、电极界面调控及抗氯离子腐蚀技术研究,多项成果发表于Adv. Energy Mater.、Small等高水平期刊,累计发表 SCI 论文 150 余篇,h 指数达 73。 ▍Email:kimsangj@jejunu.ac.kr 撰稿:《纳微快报(英文)》编辑部 编辑:《纳微快报(英文)》编辑部 关于我们 9.jpg Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc.),包括微纳米材料与结构的合成、表征、性能及其在能源、催化、环境、传感、人工智能、电磁波吸收与屏蔽、健康监测、生物医药等领域的应用研究及高水平综述。期刊已被SCI、EI、PubMed、SCOPUS等数据库收录,2025 JCR IF=38.5,学科排名Q1区前1.5%。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。 期刊网址: https://springer.com/40820 投稿网址:https://mc03.manuscriptcentral.com/nmlett E-mail: editorial_office@nmlett.org Tel: 86-21-34207624
High Polarity Doping of CoFe Layered Hydroxides: Bifunctional and Corrosion Resistant Anion Exchange Membrane Seawater Electrolyzers Anandhan Ayyappan Saj, Sampath Prabhakaran, Mohsin Rasool, Kousik Bhunia, Dongho Lee, Hyunseok Ko, Tukaram D. Dongale, Muthukumar Perumalsamy, Arul Saravanan Raaju Sundhar, Do Hwan Kim & Sang Jae Kim Nano-Micro Letters (2026)18: 393 https://doi.org/10.1007/s40820-026-02230-8 本文亮点 1. 催化机理优化:本研究采用氧化镁纳米颗粒辅助法制备钴铁层状金属氢氧化物(CoFe LMH);氟掺杂可选择性调控铁位点并保持钴位点结构完整,有效改性钴铁层状氢氧化物的电子结构,使铁原子形成高自旋构型,进而提升催化活性。 2. 抗腐蚀能力突出:本研究通过氟掺杂改性样品 F-CoFe LMH-8 为双功能催化剂,在海水环境中兼具优异的亲水性、亲氧性,同时具备显著的疏氯特性,可有效排斥氯离子。 3. 长周期运行稳定:本研究以 F-CoFe LMH-8 同时作为阴、阳极组装的阴离子交换膜电解装置,在海水电解体系中,施加 2.3 V 电压即可实现1 A⋅cm⁻²的电流密度;装置连续运行 500 小时,电压衰减率仅为0.15 μV⋅h⁻¹,展现出极佳的运行稳定性。 微信图片_2026-06-23_091114_955.png 研究背景 氢能被视作清洁能源核心载体,电解水制氢是实现 “氢经济” 的主流技术,传统电解水依赖大量淡水资源,大规模应用受限。而海水储量丰富、取用便捷,直接海水电解成为替代淡水制氢、降低成本的理想路线,但面临两大核心难题:一是海水中大量氯离子会引发电极腐蚀、发生竞争性析氯副反应,严重破坏催化剂结构、降低产氢效率;二是析氢反应(HER)与析氧反应(OER)动力学缓慢,现有非贵金属催化剂活性不足、稳定性差。 目前主流层状双氢氧化物(LDH)类电极材料,在碱性海水体系中易发生结构重构,氯离子极易侵蚀活性位点;各类阴离子插层、表面改性等手段虽有改善,但普遍存在工作电压高、稳定时长短、难以工业化的问题。而贵金属催化剂活性优异,但价格昂贵、储量稀缺,无法满足规模化海水电解需求,因此开发低成本、高活性、耐氯腐蚀的非贵金属双功能电解催化剂具有重要现实意义。 内容简介 海水电解制绿氢是一项极具发展前景的技术,但OER动力学迟缓、氯离子腐蚀等问题严重制约了其实际应用。全北国立大学Do Hwan Kim等制备了一种新型氟掺杂钴铁层状金属氢氧化物催化剂(F-CoFe LMH-8),该材料是性能优异的双功能电催化剂。在电流密度为10 mA⋅cm⁻²条件下,其HER、OER的过电位分别仅为 81.23 mV 和 265.5 mV。理论计算与实验结果表明:氟掺杂可调控材料电子结构,使铁活性位点转变为高自旋构型,优化反应中间体吸附能,同时赋予材料疏氯特性,有效抵御氯离子带来的腐蚀。将 F-CoFe LMH-8 同时作为阴、阳极双功能催化剂,组装得到阴离子交换膜水电解槽(AEMWE),在连续制氢过程中表现出优异性能:在 1 mol/L KOH溶液中,电流密度可达1.2 A⋅cm⁻²;在 1 mol/L KOH+0.5 mol/L NaCl混合溶液中,电流密度为1.02 A⋅cm⁻²;在海水基 1 mol/L KOH体系中,施加 2.3 V 电压即可实现1 A⋅cm⁻²的电流密度。此外,研究采用长短期记忆(LSTM)机器学习模型对 F-CoFe LMH-8 的稳定性进行预测。该研究思路为定向设计适用于海水电解阴离子交换膜电解槽(AEMWE)、可规模化制氢的高稳定性、疏氯型高性能电催化剂,提供了一套完整的研究方案。 图文导读 I F-CoFe LMH的制备与表征 图1展示了氟掺杂钴铁层状氢氧化物的合成流程与物化性质。本研究采用氧化镁纳米颗粒辅助法制备钴铁层状金属氢氧化物(CoFe LMH)材料,高分辨透射电镜(TEM)观测结果显示(图1b-e),氟掺杂并未破坏材料原本的纳米片形貌:其中未掺杂样品 CoFe LMH-3 的晶面间距为 0.2521 nm,氟改性后的 F-CoFe LMH-8 晶面间距增至 0.2612 nm,直观证明掺杂后材料晶格发生膨胀。结合XRD、Raman光谱与傅里叶变换红外光谱(图1f-h)测试结果可知,氟元素成功掺入晶体晶格且未生成其他杂相,同时有效调控了材料内部金属 – 氧键、羟基等化学键与表面官能团结构。图1i结合电子顺磁共振结果进一步证实,F-CoFe LMH-8 的信号强度显著高于原始样品,说明氟掺杂成功诱导铁原子形成高自旋构型,这一结构特征是该催化剂能够展现优异电催化性能的核心原因。 1.png 图1 (a) 材料合成示意图;(b、c) CoFe LMH-3 在不同放大倍数下的高分辨透射电镜图;(d、e) F-CoFe LMH-8 在不同放大倍数下的高分辨透射电镜图(插图为对应的选区电子衍射图谱);CoFe LMH-3 与 F-CoFe LMH-8 的:(f) 粉末 XRD及对应晶面间距、(g) Raman光谱、(h) 傅里叶变换红外光谱、(i) 电子顺磁共振图谱。 图2通过多种表征手段,深入分析了氟掺杂前后钴铁层状氢氧化物的元素化学态、原子配位环境与电子结构特征。XPS谱图分别采集了Co、Fe、O、F四种元素的精细谱图(图2a-d),对比测试结果可见,氟掺杂后钴元素的特征峰位置、峰形均未发生明显偏移,证明钴位点的化学价态与局域电子环境基本保持不变;而铁元素的特征衍射峰出现显著位移,结合能发生规律性改变,直观反映出氟掺杂对铁原子电子云分布产生了明显调控。由此可见氟掺杂对钴元素的电子环境影响微弱,但会显著改变铁元素的结合能,证明氟原子可选择性作用于铁位点,同时完整保留钴活性中心的结构。同时图谱中出现了清晰的氟元素特征峰,也直接证实氟原子成功负载于材料表面并参与结构构建。 在此基础上,图2e-l通过X 射线吸收近边结构(XANES)与扩展 X 射线吸收精细结构(EXAFS)测试进一步印证了这一规律:钴的 K 边吸收谱与配位键长基本无变化,而铁的 K 边吸收峰向高能方向偏移,Fe-O 键作用明显增强,金属间键合被弱化。结合小波变换扩展 X 射线吸收精细结构图谱能够看出,氟掺杂精准调控了铁的局域配位环境,最终诱导铁形成高自旋状态,从原子层面揭示了材料催化性能提升的内在机理。 2.png 图2 CoFe LMH-3 与 F-CoFe LMH-8 的 XPS 精细谱:(a-d) Co、Fe、O、F;(e) 钴 K 边 X 射线吸收近边结构谱及 (f) 对应的扩展 X 射线吸收精细结构谱;(g) 铁 K 边 X 射线吸收近边结构谱及 (h) 对应的扩展 X 射线吸收精细结构谱;钴 K 边小波变换扩展 X 射线吸收精细结构谱:(i) CoFe LMH-3、(j) F-CoFe LMH-8;铁 K 边小波变换扩展 X 射线吸收精细结构谱:(k) CoFe LMH-3、(l) F-CoFe LMH-8。 II F-CoFe LMH的电化学性能 图3综合展示了系列催化剂的电化学性能、界面反应特征与理论吸附特性。图 3a、3b分别为线性扫描伏安曲线与塔菲尔斜率,结果表明氟掺杂改性后的 F-CoFe LMH-8 拥有更优异的反应动力学性能,电荷转移阻力更低,电催化反应速率远优于未掺杂的 CoFe LMH-3。图 3c奈奎斯特图同样印证了该结论,F-CoFe LMH-8 的圆弧半径明显更小,代表材料界面电荷传输效率更高、导电性能更佳。 图 3d、3e 与 3f、3g为扫描电化学显微镜测试结果,分别呈现了两种材料的基底电流与探针电流分布。F-CoFe LMH-8 的电流响应强度显著更高,说明其表面活性位点数量丰富,且反应活性分布均匀。图 3h是 F-CoFe LMH-8 在恒定电流密度−50 mA⋅cm −2下的电化学稳定性测试,全程电位波动极小,体现出出色的电化学耐久性能。 结合理论计算,图 3i、3j、3k分析了氢吸附中间体在钴、铁、氧位点的吸附能与吉布斯自由能:铁位点对氢中间体的吸附强度最为适中,既保障反应顺利推进,又不会因吸附过强占据活性位点,从理论层面解释了该材料析氢催化活性突出的内在原因。 3.png 图3 (a) 不同组分氟掺杂与未掺杂钴铁层状氢氧化物催化剂的线性扫描伏安曲线;(b) 对应的塔菲尔斜率;(c) CoFe LMH-3 与 F-CoFe LMH-8 的奈奎斯特图;CoFe LMH-3 的扫描电化学显微镜测试:(d) 基底电流分布、(e) 探针电流分布;F-CoFe LMH-8 的扫描电化学显微镜测试:(f) 基底电流分布、(g) 探针电流分布;(h) F-CoFe LMH-8 在恒定电流密度 −50 mA⋅cm⁻² 下的电化学稳定性测试;氟掺杂钴铁层状氢氧化物催化剂上氢吸附中间体在 (i) 钴位点、(j) 铁位点、(k) 氧位点的吸附能;钴、铁、氧位点上氢吸附中间体的吉布斯自由能。 图4围绕不同组分催化剂的析氧电化学性能、反应机理及电子结构展开分析。图 4a、4b为各样品的线性扫描伏安曲线与塔菲尔斜率,对比多款未掺杂、氟掺杂钴铁层状氢氧化物可以发现,F-CoFe LMH-8 的起始电位更低、塔菲尔斜率更小,代表其OER动力学最优,催化反应速率领先其余样品。图 4c的奈奎斯特图进一步佐证了上述结论,相较于 CoFe LMH-3,F-CoFe LMH-8 的阻抗圆弧半径更小,界面电荷转移阻力更低,电子传输能力更强。图 4d、4e和图 4f、4g为扫描电化学显微镜测试结果,F-CoFe LMH-8 的基底电流与探针电流信号更强,说明材料表面活性位点分布均匀且本征活性更高。 图 4h的稳定性测试结果显示,F-CoFe LMH-8 在持续工作过程中性能保持稳定,具备良好的长效服役能力。结合图 4iOER机理图与图 4j吉布斯自由能图,可明确材料的反应路径与最优活性位点。图 4k、4l的分态密度图谱则揭示了电子结构差异:氟掺杂有效调控了费米能级附近的电子态密度,提升了电子迁移能力,最终从电子层面阐明了 F-CoFe LMH-8 高效催化OER的本质。 4.png 图4 (a) 多种样品的线性扫描伏安曲线;(b) CLMH、CoFe LMH-1、CoFe LMH-2、CoFe LMH-3、CoFe LMH-4、FLMH-5、F-CLMH-6、F-CoFe LMH-6、F-CoFe LMH-7、F-CoFe LMH-8、F-CoFe LMH-9 及 F-FLMH-10 的塔菲尔斜率;(c) CoFe LMH-3 与 F-CoFe LMH-8 的奈奎斯特图;CoFe LMH-3 的扫描电化学显微镜测试:(d) 基底电流、(e) 探针电流;F-CoFe LMH-8 的扫描电化学显微镜测试:(f) 基底电流、(g) 探针电流;(h) F-CoFe LMH-8 的稳定性测试;(i) OER机理;(j) F-CoFe LMH OER的吉布斯自由能图(用于确定反应活性位点);(k) CoFe LMH-3、(l) F-CoFe LMH-8 不同配位位点的分态密度图。 III F-CoFe LMH的耐蚀性测试与机理 图5聚焦催化剂在海水体系下的耐腐蚀性、电化学行为与作用机制。图 5a测试了 F-CoFe LMH-8 在纯KOH溶液、模拟海水以及添加天然海水的电解液中的线性扫描伏安曲线,样品在三类体系中均保持稳定的电化学响应,初步体现出良好的海水环境适应性。图 5b为两种材料在模拟海水中的线性极化曲线,相比 CoFe LMH-3,F-CoFe LMH-8 的腐蚀电流更低,抗腐蚀能力大幅提升。图 5c氯离子吸附能图谱直观说明,F-CoFe LMH-8 对氯离子的吸附作用较弱,能够有效减少氯离子在材料表面附着,这也是其具备优异疏氯性能的关键。图 5d奈奎斯特图与图 5e弛豫时间分布分析结果显示,氟掺杂改性后材料的阻抗特征更优,界面结构在模拟海水中不易被破坏。图 5f、5g多电位相频特性曲线进一步反映出,F-CoFe LMH-8 在不同电位下电化学界面状态更稳定。 图 5h原位拉曼光谱证明,长期处于模拟海水环境中,材料的晶体结构与化学键未发生明显改变,结构稳定性优异;图 5i长效稳定性测试也验证了该结果。图 5j–5q为不同电位下的扫描电化学显微镜测试数据,在 0.3 V 和 0.35 V(vs Ag/AgCl)电位条件下,F-CoFe LMH-8 的基底电流与探针电流信号始终稳定且强度更高,表明其表面活性位点在海水体系中可正常发挥作用。最后,图 5r为离子作用机理示意图,清晰阐释了材料排斥氯离子、保障催化反应持续进行的微观原理。 5.png 图5 (a) F-CoFe LMH-8 在 1 mol/L KOH溶液、模拟海水(1 mol/L KOH+0.5 mol/L NaCl)以及天然海水配制的 1 mol/L KOH 溶液中的线性扫描伏安曲线;(b) CoFe LMH-3 与 F-CoFe LMH-8 在模拟海水中的线性极化曲线;(c) 氯离子吸附能图谱;(d) 奈奎斯特图;(e) CoFe LMH-3 与 F-CoFe LMH-8 的弛豫时间分布分析;(f) CoFe LMH-3、(g) F-CoFe LMH-8 的多电位相频特性分析;(h) F-CoFe LMH-8 在模拟海水中的原位拉曼测试结果;(i) 其电化学稳定性测试结果。在参比电极银 / 氯化银电位为 0.3 V 条件下,CoFe LMH-3 的扫描电化学显微镜测试:(j) 基底电流、(k) 探针电流;电位升至 0.35 V 时:(l) 基底电流、(m) 探针电流。在 0.3 V(vs Ag/AgCl)条件下,F-CoFe LMH-8 的扫描电化学显微镜测试:(n) 基底电流、(o) 探针电流;电位升至 0.35 V 时:(p) 基底电流、(q) 探针电流。(r) 离子在 F-CoFe LMH-8 材料表面的作用机理示意图。 IV F-CoFe LMH的稳定性测试 图6主要探究了催化剂在电解液及电解器件中的综合性能、长效稳定性与运行表现。图 6a、6b是 CoFe LMH-3 与 F-CoFe LMH-8 在模拟海水电解液(1 mol/L KOH+0.5 mol/L NaCl)中的线性扫描伏安曲线与多阶电流密度稳定性测试,对比可见 F-CoFe LMH-8 电化学活性更出色,在阶梯式变化的电流工况下性能也能平稳维持。图 6c进一步开展大电流稳定性测试,该样品在200 mA⋅cm⁻²、400 mA⋅cm⁻²高电流密度下持续工作,电位无明显波动,耐受大电流冲击的能力较强。 图 6d为阴离子交换膜水电解槽(AEMWE)装置示意图,直观展示了器件整体结构。图 6e、6f分别是搭载 F-CoFe LMH-8 双功能催化剂的电解槽,在三种不同电解液体系下的极化曲线与奈奎斯特图,结果表明该电解槽在常规碱液、模拟海水、天然海水体系中均拥有较低的极化电压与界面阻抗,环境适配性良好。 图 6g记录了电解槽的长期运行稳定性,器件可连续稳定工作。图 6h将电解槽电压衰减率与美国能源部(DOE)既定指标进行对比,本体系衰减速率远低于标准要求,长效运行优势显著。图 6i针对海水体系下的电解槽开展不同运行时长的弛豫时间分布分析,数据证明随着运行时间增加,器件界面结构与电化学特性未出现明显劣化。图 6j借助时序分析法对催化剂及电解器件的长期稳定性进行预测,进一步佐证了该材料在海水电解规模化应用中的潜力。 6.png 图 6 (a) CoFe LMH-3 与 F-CoFe LMH-8 催化剂在 1 mol/L KOH+0.5 mol/L NaCl体系中的线性扫描伏安曲线;(b) 多阶电流密度下的稳定性测试结果;(c) F-CoFe LMH-8 在上述电解液中,分别恒定电流密度为 200 mA⋅cm⁻²、400 mA⋅cm⁻² 时的稳定性测试;(d) 阴离子交换膜水电解槽(AEMWE)装置示意图;(e) 采用 F-CoFe LMH-8 双功能催化剂组装的电解槽,分别在 1 mol/L KOH、1 mol/L KOH+0.5 mol/L NaCl、1 mol/L KOH+天然海水体系下的极化曲线;(f) 对应的奈奎斯特图;(g) 该电解槽的长期运行稳定性;(h) 电压衰减率与美国能源部(DOE)技术指标对比;(i) F-CoFe LMH-8 基电解槽在海水体系中不同运行时长下的弛豫时间分布分析;(j) 借助时序分析法对催化剂稳定性进行的预测结果。 V 总结 综上所述,本研究对钴铁层状金属氢氧化物进行氟掺杂改性,成功制备出一种性能优异、稳定性强的双功能电催化剂,可应用于长效海水电解体系。氟掺杂可精准调控钴铁层状氢氧化物的铁配位位点,同时不会破坏用于OER的钴活性金属中心。所制备的 F-CoFe LMH-8 兼具出色的析氢与析氧催化性能:电流密度达到10 mA⋅cm⁻²时,析氢过电位为 81.23 mV,析氧过电位为 265.5 mV。氟掺杂能够强化铁位点的铁 – 氧键作用,促使双电层内氢氧根离子持续富集,有效抑制氯离子腐蚀,进而提升催化剂的催化活性与服役寿命。此外,采用 F-CoFe LMH-8 同时作为阴、阳极组装的阴离子交换膜海水电解槽,在 2.3 V 电压下可实现1 A⋅cm⁻²的电流密度;在0.125 A⋅cm⁻²恒定电流下连续运行 500 小时,电压衰减率仅为0.15 μV⋅h⁻¹,不仅达到美国能源部相关技术指标,也证明该体系具备工业化应用潜力。本研究为简易制备高性能、抗腐蚀海水电解催化剂提供了新思路,可推动阴离子交换膜海水电解槽的长效稳定运行。 作者简介 7.jpg图片 Do Hwan Kim 本文通讯作者 韩国全北国立大学 教授 ▍主要研究领域 电催化水分解、海水电解、阴离子交换膜电解槽技术以及非贵金属催化材料设计。 ▍主要研究成果 Do Hwan Kim教授任职于韩国全北国立大学,为科学教育学部教授、表面化学实验室负责人,主要研究方向涵盖电催化水分解、海水电解、阴离子交换膜电解槽技术以及非贵金属催化材料设计,在Adv. Funct. Mater.、Nano Energy 等知名期刊累计发表 SCI 论文 130 余篇,总引用量超 5000 次,h 指数约 40,研究成果多见于,在本研究中主要负责催化剂设计、电子结构调控与电解槽性能测试分析。 ▍Email:dhk201@jbnu.ac.kr 图片8.png Sang Jae Kim 本文通讯作者 韩国济州国立大学 教授 ▍主要研究领域 海水电解双功能催化剂、层状氢氧化物复合材料、电极界面调控及抗氯离子腐蚀技术。 ▍主要研究成果 Sang Jae Kim教授毕业于日本东北大学,曾赴剑桥大学、佐治亚理工学院开展访学研究,现任韩国济州国立大学教授、纳米材料与系统实验室负责人,长期致力于海水电解双功能催化剂、层状氢氧化物复合材料、电极界面调控及抗氯离子腐蚀技术研究,多项成果发表于Adv. Energy Mater.、Small等高水平期刊,累计发表 SCI 论文 150 余篇,h 指数达 73。 ▍Email:kimsangj@jejunu.ac.kr 撰稿:《纳微快报(英文)》编辑部 编辑:《纳微快报(英文)》编辑部 关于我们 9.jpg Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc.),包括微纳米材料与结构的合成、表征、性能及其在能源、催化、环境、传感、人工智能、电磁波吸收与屏蔽、健康监测、生物医药等领域的应用研究及高水平综述。期刊已被SCI、EI、PubMed、SCOPUS等数据库收录,2025 JCR IF=38.5,学科排名Q1区前1.5%。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。 期刊网址: https://springer.com/40820 投稿网址:https://mc03.manuscriptcentral.com/nmlett E-mail: editorial_office@nmlett.org Tel: 86-21-34207624 过程所郭旸旸/姚明水/朱廷钰等综述:柔性MOFs在CO₂及其同位素中的吸附分离机制、调控策略与潜在应用
过程所郭旸旸/姚明水/朱廷钰等综述:柔性MOFs在CO₂及其同位素中的吸附分离机制、调控策略与潜在应用