研究背景
将太阳能转化为化学能的光催化技术,被视为一种有前途的可持续能源转化手段。石墨相氮化碳(g-C₃N₄)作为一种环保的非金属光催化剂,自发现可用于光催化产氢以来,引起了研究者们广泛的关注。相较于传统的TiO₂材料,g-C₃N₄的光催化活性、稳定性更为优越。然而,其本体材料面临的可见光吸收不足、光生载流子传输动力缓慢、空穴电子在体相、表面的严重复合,较大程度地制约了其在水分解制氢(HER)、二氧化碳还原反应(CRR)、氮气还原反应(NRR)、光阴极保护(PCP)、污染物去除以及氧析出反应(OER)等方面的应用,在其大规模应用之前,仍面临诸多挑战。
Decade Milestone Advancement of Defect-Engineered g-C₃N₄ for Solar Catalytic Applications
Shaoqi Hou, Xiaochun Gao*, Xingyue Lv, Yilin Zhao, Xitao Yin, Ying Liu, Juan Fang, Xingxing Yu, Xiaoguang Ma*, Tianyi Ma* & Dawei Su*
Nano-Micro Letters (2024)16: 70
https://doi.org/10.1007/s40820-023-01297-x
本文亮点
1. 总结了近十年来,缺陷工程策略在提升g-C₃N₄光催化活性的重要进展,并强调了结晶调节和缺陷态构建对实现更精确的缺陷化g-C₃N₄材料“定制”的重要作用。
2. 探讨了缺陷能级在g-C₃N₄光催化活性的作用,尤其是利用飞秒瞬态吸收光谱技术揭示缺陷态对光生载流子转移动力学的影响。
3. 展望了精确“定制”缺陷化g-C₃N₄的前景。
内容简介
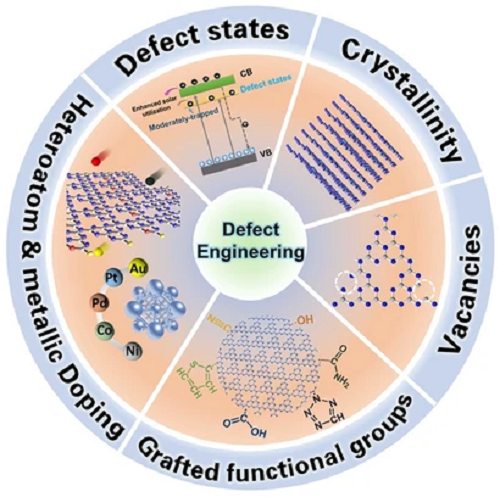
近十年,g-C₃N₄在各个光催化领域,已成为炙手可热的光催化剂。但由于其较差的太阳能捕获能力和缓慢的电荷转移动力学,其仍然面临光生载流子供应不足的问题。研究人员发现,缺陷工程策略可以在改进g-C₃N₄纳米结构特性的同时,优化其电子能带结构,进而显著缓解上述问题。为此,澳大利亚悉尼科技大学苏大为等对缺陷工程策略进行了全面讨论,包括利用空位/非金属掺杂手段来优化g-C₃N₄电子能带结构和电子密度、利用金属掺杂获得超活性配位环境(M-N ₓ, M-C₂N₂, M-O键)、使用官能团接枝来优化其能带结构,以及削弱层间范德华相互作用促进结晶度获得扩展的共轭π体系。其中,强调了N空位、P/S/卤素掺杂、氰基等缺陷类型引起的缺陷态在促进太阳能收集和光载流子转移的重要作用。更重要地,综述了飞秒瞬态吸收光谱(fs-TAS)在浅缺陷态能级识别与载流子寿命分析的最新进展。我们相信这篇综述可以引发读者对“定制”缺陷化g-C₃N₄课题进行深层次的思考,并推动基于g-C₃N₄的光催化技术取得更为丰富的成果。
图文导读
I 缺陷工程
通过缺陷工程,在g-C₃N₄基体中引入杂质或者改变原子排列周期性,可有效改善其电子能带结构、光学性质和导电性。有趣的是,制备缺陷化g-C₃N₄的过程中,可调控前驱体类型、反应模板和退火条件(热解气氛、加热速率、退火时间和压力)进一步优化g-C₃N₄的外在形貌。因此,具有缺陷的g-C₃N₄样品通常表现出扩展的太阳能收集能力、高效的光载流子转移过程以及更大比表面积和更多活性位点,从而提高其光催活性。
1.1缺陷工程的设计原理
一般来说,g-C₃N₄的缺陷工程应遵循三个重要原则,即基本建立丰富的活性位点、增强太阳能收集能力和光生载流子的高效传输(图1)。为了实现具有最佳性能的缺陷g-C₃N₄的最终目标,还需要开展更多的研究工作,包括精确控制结晶度、浅缺陷态甚至优化表面态。要实现缺陷定制,还需要更先进的原位探测技术,如原位漫反射红外傅立叶变换光谱(DRIFTS)和原位飞秒瞬态光谱技术。
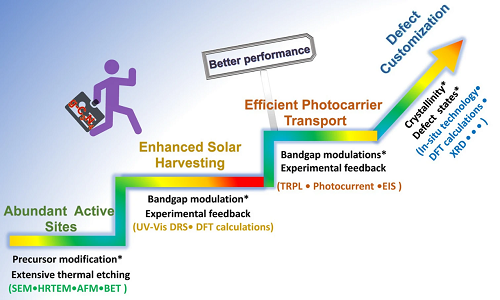
图1. 优化g-C₃N₄光催化性能的设计原理。
1.2空位(C/N)
通过改变合成条件,可以获得含有C空位或N空位的g-C₃N₄,其中的空位类型和位置可以通过C或N的电子顺磁共振(EPR)信号和解析X射线光电子能谱(XPS)峰面积比来确定。一般来说,C和N两种缺陷都能赋予g-C₃N₄优化的电子结构,包括更窄的带隙、更强的太阳能光吸收、更有利的电荷分离和传输,进而提高太阳能利用率(图2a)。
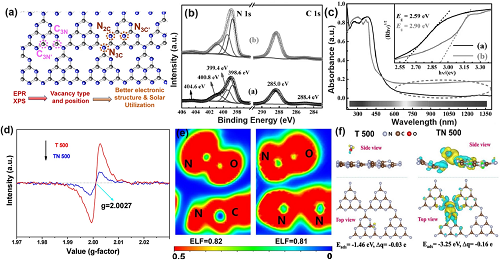
图2. C3N 空位的缺陷控制。
1.3非金属掺杂
与C和N空位类似,对g-C₃N₄进行非金属掺杂(如C、P、S、O、B和F等杂原子),同样可以优化g-C₃N₄的电子结构,并提高其可见光吸收和电荷分离效率。
例如研究人员通过直接在空气中对三聚氰胺进行退火实验成功合成了聚合的g-C₃N₄(PCN)。根据以氨基胍/尿素为前驱体制备的N掺杂量较低化合物命名为APCN,以及以尿素/氨基胍/三聚氰胺为前驱体制备的N掺杂量较高化合物命名为NPCN,我们观察到光催化HER率逐渐提高,分别达到5.81、6.97和40.32mmol g⁻1。根据PCN<APCN<NPCN的递增顺序,我们看到光催化HER率逐渐提高,分别为5.81、6.97和40.32 mmol g⁻1。随后,作者给出了三个计算模型,从能带结构、电荷密度分布以及ΔG变化等方面模拟了不同N掺杂浓度的g-C₃N₄(图 3)。与PCN相比,NPCN在禁带中显示出新的缺陷能级(也称为缺陷态或者mig-gap states),这主要是由于存在新的N掺杂所致(图3 a-d)。APCN的情况也是如此。作者称,这可能更有利于光电载流子从这些缺陷态和CBM中分离出来。电子密度图还显示有更多的电子从N掺杂物转移到H原子上,从而进一步加速了光载流子转移动力学(图3 e)。
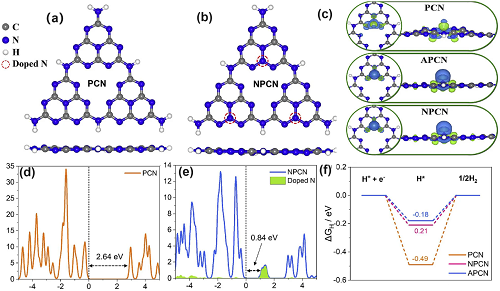
图3. N掺杂的缺陷控制。
II 金属掺杂
一般而言,金属掺杂剂通常能够增强g-C₃N₄对太阳光的吸收能力,并提高电子转移和光载流子分离效率。金属掺杂通常通过将g-C₃N₄前驱体与可溶性金属盐混合物进行热解来实现。通过球差电镜和K-边X射线吸收精细结构(EXAFS)等技术可以在原子水平上区分不同形态的金属,并识别具有相互作用和键合物质环境下的金属配位。
2.1碱金属掺杂
在g-C₃N₄的电子能带结构和光学性能方面,研究发现K和Na掺杂对其产生不同影响。理论研究发现K和Na原子均可缩小g-C₃N₄的带隙,并增强其对太阳光吸收的能力。此外,K原子更倾向于出现在层间空间,为电子提供更好的垂直转移途径;而Na原子则倾向于与平面内的N原子形成化学键,因为Na 3s电子容易逃逸。实验结果表明,相较于Na掺杂,掺入K元素使得g-C₃N₄具有更高的NO光降解速率。此外,其他碱金属如Ba和Rb对于促进g-C₃N₄的太阳活性也具有有效作用。
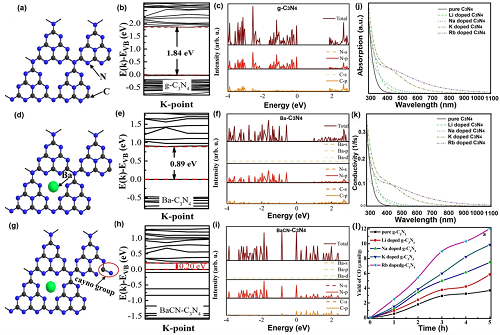
图4. 碱金属掺杂的缺陷控制。
2.2过渡金属掺杂
过渡金属如Co、Cu、Fe、Ce和Bi对g-C₃N₄也可提升g-C₃N₄光催化活性。最近,金属单原子(M-SAs)在水裂解、CO₂还原等电化学催化中展现出稳定性,具有良好的应用前景。此后,更多的研究关注于M-SAs掺杂g-C₃N₄体系,包括确定金属-氮(M-N)、金属-氧的相互作用、形成双原子催化剂甚至单原子-金属簇催化剂(图5a)。

图5. 金属单原子掺杂的g-C₃N₄的缺陷控制。
III 官能团
将有机官能团接枝到g-C₃N₄上是最具潜力的缺陷控制方法之一,可通过优化能带结构、增强太阳吸收以及提高光载流子输运速率来调整g-C₃N₄的催化性质。先前报道中所涉及的官能团包括氰基(-C≡N)、脲类基团(O=C-NH₂)、羧基(-COOH)、酮基(-C=O)、羟基(-OH)和各种芳环,其中研究最广泛的是氰基。
3.1氰基(-C≡N)
研究发现通过双氰胺和KOH的退火和后续超声处理,制备了具有-C≡N接枝的g-C₃N₄纳米带(mCNN)(图6a)。根据傅立叶变换红外光谱(图6b),mCNN中的-C≡N键在2150 cm⁻1附近呈现出典型的吸收峰,而XANES光谱中的N K-边在406.3eV附近显示宽峰,表明电子从N 1s转移到了C-N σ*轨道。UV-VIS DRS证实,相较于原始g-C₃N₄,mCNN对太阳光的吸收范围更广,在约450nm处出现明显肩峰。此外,mCNN的吸收尾迹归因于-C≡N诱导的缺陷态,这将其太阳光吸收范围扩大到700 nm,意味着太阳能利用率显著提高。因此,与纯g-C₃N₄相比,mCNN在氮气和氩气环境下的NH3产率都有所提高。
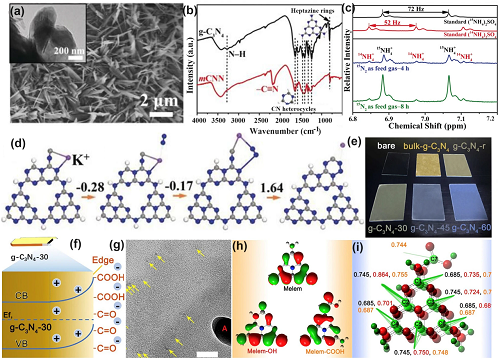
图6.氰基官能团的g-C₃N₄缺陷控制。
3.2含氧官能团
枝接含O官能团,如-COOH、-C=O和-OH也可提高g-C₃N₄的太阳能利用率。研究发现通过无机酸氧化法可制备带有-COOH和-C=O基团的g-C₃N₄,随着氧化蚀刻时间的延长,g-C₃N₄的颜色由黄色变为蓝色,表明含O基团的种类增多,带隙增大(图 6e),有利于建立优化的电子传输通道。与原始g-C₃N₄和还原g-C₃N₄-30样品相比,电子流中的电荷耗尽层更厚,能带更弯曲(图 6f,g)。HOMO图表明,电子容易聚集在O原子上,而相邻C原子周围的电子密度较低(图 6h)。同样,含O基团修饰的g-C₃N₄的C原子边缘的正电荷量远高于远离边缘的C原子或原始g-C₃N₄的C原子,这进一步表明电子倾向于聚集在g-C₃N₄-30纳米片的边缘(图 6i)。作者认为,这可以加速电荷分离,缩小带隙,促进H₂O₂的生成,从而达到除菌的目的。
3.3芳环基团
通过冷冻干燥退火处理尿素和3-氨基-1,2,4-三氮唑(3-AT)的混合物成功地获得了具有三唑基团和-C≡N基团的多孔氰酰胺-三唑-七嗪聚合物(CTHPx, x为尿素与3-AT的质量比)。原始g-C₃N₄(CN)、三唑基改性CN(THP)和氰酰胺基改性CN (CHP)被用作对照样品。通过FT-IR光谱分析确定CTHP30中存在着三唑基团和-C≡N基团,它们在3400、2175和739 cm⁻1左右显示出典型峰位,分别归属于-NH₂、-C≡N和N-N (图7a)。图7b的固态核磁共振光谱则详细分析了各个峰位之间的关系。根据紫外-可见DRS,我们可以观察到这些三唑基团或-C≡N改性g-C₃N₄的太阳吸收增强,其中不含氰基团的THP的可见光吸收最好,其次是CTHP30 (图7c)。
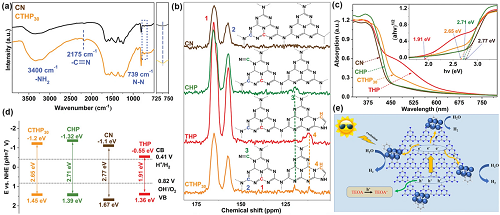
图7. 芳环基团的缺陷控制。
IV 高结晶性的g-C₃N₄
与g-C₃N₄的缺陷构筑工程相反,较高的结晶度通常表明其原子排列更加规则,具有扩展的共轭结构,从而稳定了π电子系统以实现快速电荷迁移,并由于减少了带隙和缺陷引起的光电载流子陷阱而提高了太阳能利用率。其中,正文给出同时调节高结晶g-C₃N₄和建立缺陷两种手段,共同提高光催化活性的典型实例。
PHI型g-C₃N₄(PHI-CN)是目前研究中广泛探讨的模型,其由无限重复的三-s-三嗪单元构成(图8a)。而PTI型g-C₃N₄(PTI-CN)则由桥接位点上的氮原子连接三嗪单元形成(图8b)。一般来说,PHI-CN通常通过简单的离子热策略合成,使用大块g-C₃N₄和MCl (M=Li,Na,K)作为前驱体。与PTI型g-C₃N₄ (PTI-CN)相比,大多数PHI型g-C₃N₄(PHI-CN)材料都表现出明显改善光催化性能。
4.1聚七嗪酰亚胺(PHI)
研究发现PHI-CN的计算带隙比PTI-CN小(1.17对3.23 eV),这是因为扩展的共轭π系统(图 8c-d),PHI-CN具有卓越的可见光响应能力。此外,PTI-CN中夹杂的Li⁺和Cl⁻对缩小禁带宽度的作用很小。在XRD图谱中,分配给g-CN-1的PHI-CN显示出更高的结晶度,其(002)和(001)峰更尖锐,与块体对应物的峰值方向相反。这可能是由于氢键较少,层间化学作用较强,从而提高了聚合度。由于结构和结晶度的差异,g-CN-1的光催化HER速率最高,达到770 μmol h⁻1,远远超过PTI/Li⁺Cl⁻,这表明PHI单元比PTI单元更具优势。

图8. 高结晶性材料的缺陷控制(PHI)。
4.2聚三嗪酰亚胺(PTI)
离子热法可获得PTI基碳氮化物,该方法采用氯化锂和氯化钾的共晶混合物作为溶剂。该合成路线分为两个步骤:(1)在惰性氩气氛下,将双氰胺和熔盐在400-500 ℃下预热;(2) 将上述混合物在真空中长时间退火至48小时,得到褐色PTI/Li⁺Cl⁻。
通过XRD、TEM和固体核磁共振谱分析发现,PTI/Li⁺Cl⁻具有较高的结晶度,并通过微弱的范德华力分离。此外,Li⁺和Cl⁻位于沿z轴方向的通道中(图9a)。进一步对剥离后的超薄PTI纳米片进行研究发现,HRTEM图像显示其六角形和三嗪单元保持不变(图9b-c)。有趣的是,由此产生的PTI纳米片能够在太阳照射下产氢(图9d)。然而,在TEOA溶液中它的循环HER活性遭受了严重衰减,表明其在碱性环境中不稳定。改用甲醇添加剂后,在130小时内其HER性能显示出良好稳定性,表明其具有广阔的光催化应用前景。
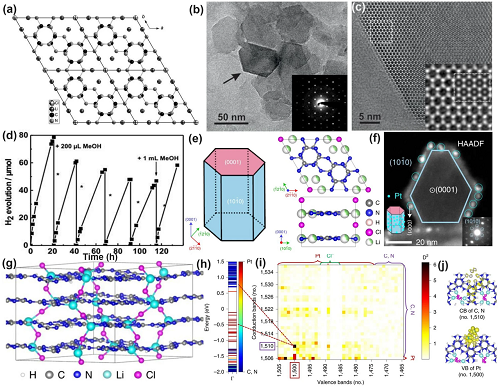
图9. 高结晶性材料的缺陷控制(PTI)。
4.3关于fs-TAS缺陷陷阱的最新讨论
近年来,出现了利用飞秒瞬态吸收光谱仪(fs-TASM)进行研究的方法,来识别浅缺陷态,并探索深层光载流子转移动力学的研究。典型的fs-TASM系统具有时间和空间分辨能力,由一个产生数十飞秒脉冲的飞秒Ti/蓝宝石再生放大激光系统和一个数据采集瞬态吸收光谱仪组成。通过放大器和BBO晶体后,可以得到泵浦激光器和白光连续光谱作为探测脉冲。将泵浦光束和探测光束聚焦在样品上,实现时间和空间重叠(图10a)。
最近有研究制备了B和P共掺杂的g-C₃N₄(BPCN),并根据DFT计算发现电子可以沿着P→N→C→B的路径转移(图10b)。此外,在最小ΔG变化为0.16 eV时,进一步表明BPCN更有利于活性H*的吸收和解吸过程,并提高HER性能(图10c)。利用飞秒瞬态吸收光谱(fs-TAS)揭示了深层电荷转移动力学,具体来说,在原始BCN中只观察到负信号范围在420-800 nm之间,主要是因为受激发射(SE,图10d-e)引起的。然而,BPCN在420-640 nm和640-800 nm范围内分别出现了负信号和正信号(图10g-h)。BPCN最强正吸收带归因于由光电子诱导引起的激发态吸收(ESA),表明快速电荷激发与分离过程。
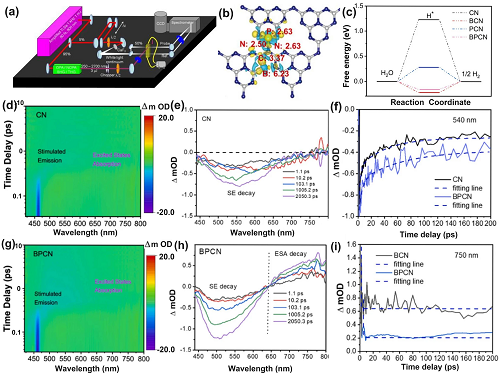
图10. 关于fs-TAS缺陷陷阱的最新讨论。
V 总结与展望
在过去的10年中,缺陷工程g-C₃N₄获得了巨大的发展,通过优化电子结构、电子电导率和电子极化,提高了其在光收集和电荷转移动力学方面的太阳能利用率。我们重点介绍了空位产生、杂原子和金属原子掺杂杂质、接枝官能团的缺陷修饰以及结晶度控制等调节策略。尽管缺陷工程g-C₃N₄的研究取得了长足的进步,但未来在进一步调节缺陷相关能级、控制缺陷浓度、加强缺陷稳定性等方面仍有突破的空间。
作者简介

本文第一作者
基于g-C₃N₄ 材料的光催化和光阴极保护。

本文通讯作者
基于 g-C₃N₄ 材料的光催化转化和等离子催化转化、金属硫电池等。
▍Email:Xiaochun.Gao@ldu.edu.cn

本文通讯作者
电子碰撞、光离子化过程和等离子体催化技术。

本文通讯作者
先进能源存储和转换的纳米结构电极材料的理论计算、合成和表征,包括锂离子电池、钠离子电池、锂硫电池和锂空气电池。
▍Email:Dawei.Su@uts.edu.au
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 悉尼科技大学苏大为等综述:缺陷化g-C₃N₄在光催化应用研究中的里程碑进展