研究背景
利用铜基催化剂上的电化学还原CO₂反应(CO₂RR)将CO₂转化为高价值燃料和化学品是一种很有前景和吸引力的CO₂转化利用技术。各种碳负载单原子催化剂(如Ni,Fe,Co和Zn)在CO₂RR中表现出良好的CO₂-to-CO性能,引起了对于Cu单原子催化剂(Cu-SACs)上的CO₂RR研究的关注和兴趣。寻找合适的催化剂载体,以将活性单原子锚定在一个稳定和良好配位环境中,成为制备结构明确且稳定的Cu-SACs的关键。石墨相氮化碳(g-C₃N₄)具有周期性的均三嗪结构和由均三嗪单元之间的6对具有孤对电子的吡啶氮原子组成的结构明确的“氮空穴”,有利于实现高密度金属原子的嵌入和提供明确的配位环境,成为锚定金属单原子的理想载体。本文提出一种简便的热聚合的策略合成了g-C₃N₄负载的Cu SACs催化剂,通过Cu含量的改变实现了Cu位点密度和Cu配位环境的调控。在CO₂RR中,优化的Cu₀.₀₅-CN催化剂表现出最高的CH₄法拉第效率达到49.04 %,CH₄电流密度7.97 mA/cm2,以及超过9的甲烷/乙烯之比(-1.2 VRHE)。结合材料结构分析、CO₂RR性能和DFT计算分析,提出嵌入g-C₃N₄氮空穴的Cu原子与氮空穴中的N原子配位形成主要的Cu⁺位点是高效的CO₂RR-to-CH₄活性中心。
Atomic Cu sites engineering enables efficient CO₂ electroreduction to methane with high CH₄/C₂H₄ ratio
https://doi.org/10.1007/s40820-023-01188-1
本文亮点
1. 采用原位热聚合的策略合成了Cu掺杂的石墨化氮化碳(g-C₃N₄)材料,通过调节Cu的掺杂水平,实现Cu在g-C₃N₄上的原子级分散和配位结构调控。
2. g-C₃N₄负载的Cu单原子催化剂上首次实现了CH₄的高法拉第效率(49.04%)和高CH₄/C₂H₄比(35.03)。
3. 基于实验和理论研究的构效关系分析表明,嵌入g-C₃N₄的氮空穴中Cu单原子与N原子配位,成为CO₂电化学还原为CH₄的高效活性位点。
内容简介
利用铜基催化剂电化学还原CO₂(CO₂RR)为高价值燃料和化学品是一种很有前景和吸引力的CO₂转化利用技术。东华大学杨建平研究员课题组采用原位热聚合的策略合成了Cu掺杂的石墨化氮化碳(g-C₃N₄)材料,通过调节Cu的掺杂水平,实现Cu在g-C₃N₄上的原子级分散和配位结构调控。首次在g-C₃N₄负载的Cu单原子催化剂上首次实现了CH₄的高法拉第效率(49.04 %)和高CH₄/C₂H₄比(35.03),基于实验和理论研究的构效关系分析表明,嵌入g-C₃N₄的氮空穴中Cu单原子与N原子配位,成为CO₂电化学还原为CH₄的高效活性位点。
图文导读
I Cuₓ-CN催化剂的合成与结构表征
首先表征了催化剂的形貌和Cu物种的分散状态。透射电子显微镜(TEM)和高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像证实了CN和Cuₓ-CN催化剂的堆叠层状结构,并排除了这些催化剂中结晶或聚集的Cu物种的存在。通过球差校正的HAADF-STEM,在Cu₀.₀₅-CN催化剂上观察到亮点分散分布在g-C₃N₄载体上,表明催化剂中孤立的Cu单原子。此外,这一简单的策略可以推广到其他过渡金属,形成g-C₃N₄支持的单金属原子,如Ni和Fe。
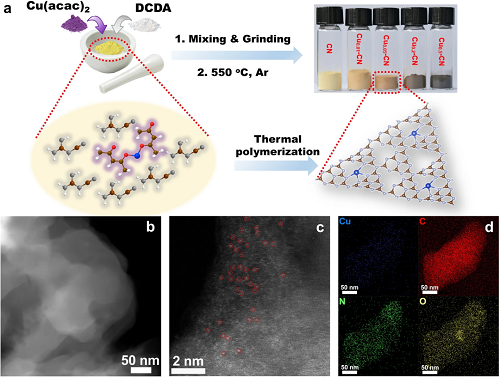
图1.Cuₓ-CN催化剂的合成示意图和Cuₓ-CN催化剂的光学照片(a);Cu₀.₀₅-CN催化剂的HAADF-STEM图像(b);像差校正的HAADF-STEM图像(c)和EDS mapping (d)。
XRD、FT-IR和13C NMR结果明确表明,低Cu负载的Cu₀.₀₁-CN和Cu₀.₀₅-CN催化剂很好地保留了g-C₃N₄主体的基本结构单元和框架,这可能与具有Cu-N结构的g-C₃N₄的氮腔中优先掺入孤立的Cu原子有关。另一方面,Cu₀.₂-CN和Cu₀.₅-CN催化剂的C-N杂环和g-C₃N₄框架结构的破坏表明,在相对较高的铜负载下,原子配位发生了变化。
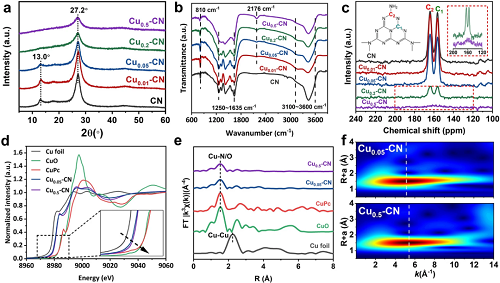
图2. (a) Cuₓ-CN催化剂的XRD图谱;(b) Cuₓ-CN催化剂的FT-IR光谱。(c) Cuₓ-CN催化剂的固态13C NMR谱;(d) Cu箔、Cu₀.₀₅-CN、 Cu₀.₅-CN、 CuPc和CuO样品的Cu K-edge XANES光谱;(e)傅立叶变换(FT) EXAFS光谱,Cu箔、Cu₀.₀₅-CN、Cu₀.₅-CN、CuPc和CuO样品的Cu K-edge;(f) Cu₀.₀₅-CN和Cu₀.₅-CN催化剂的小波变换(WT)。
通过XPS光谱分析了CN和Cuₓ-CN催化剂中元素的键合和化学状态。图3f-h总结了Cu 2p、C 1s和N 1s光谱的拟合结果。根据图2中催化剂的结构表征,拟合结果表明,对于Cu₀.₀₁-CN和Cu₀.₀₅-CN催化剂,C和N的种类和比例几乎不受影响。结合XAFS测量结果,这两种催化剂中分离的Cu原子优先嵌入氮空穴形成Cu-N配位结构,对C-N杂环结构影响较小。然而,随着催化剂中Cu含量的增加,显著降低的N(1)和C(2)峰进一步证实了在Cu₀.₂-CN和Cu₀.₅-CN催化剂中g-C₃N₄载体的C-N杂环结构的破坏。同时,Cu₀.₂-CN和Cu₀.₅-CN催化剂中Cu(2)物种和O原子比的同时增加表明,Cu2⁺物种的出现与C-O结构的增加有关,这可以从增强的C(3)峰和O 1s峰得到证明(图3g)。
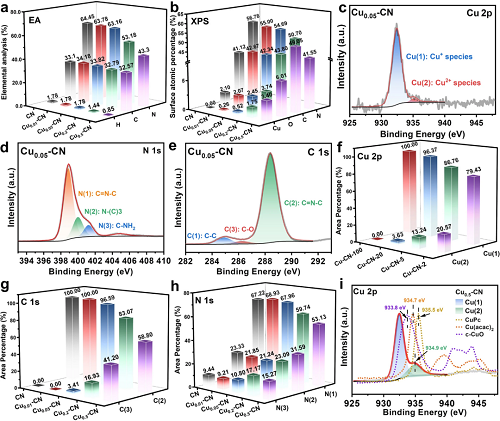
图3. (a)催化剂元素分析;(b)通过XPS测定的催化剂的表面组成;(c-e) Cu₀.₀₅-CN催化剂的Cu 2p、N 1s和C 1s谱峰拟合;(f-h)不同催化剂的Cu 2p、C 1s和N 1s谱峰拟合总结;(i) Cu₀.₅-CN催化剂与其他铜化合物的Cu 2p光谱的比较。
II Cuₓ-CN催化剂的CO₂RR催化性能
在H-cell中,以0.1 M KHCO₃作为电解质,评估了CN和Cuₓ-CN催化剂的CO₂RR性能。在Cu₀.₀₅-CN催化剂上,CH₄电流密度为7.97 mA/cm2,在-1.2 VRHE时最高FE值为49.04 %,在-1.3 VRHE时进一步增加到9.78 mA/cm2(图4c-d)。据报道,在CO₂RR中,孤立的Cu位点倾向于将CO₂转化为C1产物,如CO、CH₄、CH₃OH。另一方面,如果原子分散的铜在反应条件下可逆地转化为团簇或纳米粒子,或者有额外的活性中心与单个铜位点协同作用,也可以在铜单原子催化剂上得到C2+产物。鉴于Cuₓ-CN催化剂中存在两种可调控和可调节的Cu位点,本文比较了不同电位下Cuₓ-CN催化剂上CH₄/C₂H₄的FE比。如图4f所示,CH₄/C₂H₄的FE比随着电势的降低而增加。Cu含量最低且只有单原子Cu位点的Cu₀.₀₁-CN催化剂在-1.3 VRHE时,CH₄/C₂H₄的FE比高达35.03。这样高的CH₄/C₂H₄的FE比大多数报道的结果都要好,这是由于氮空穴中孤立的Cu原子位点对CH₄的高选择性,以及低Cu负载的Cu₀.₀₁-CN催化剂中这些位点之间的较大距离。在-1.2 VRHE条件下,Cu₀.₀₅-CN催化剂的CH₄的FE最高,同时,CH₄/C₂H₄比高达9.03。因此,Cu₀.₀₁-CN催化剂上CH₄/C₂H₄的高FE比和Cu₀.₀₅-CN催化剂上CO₂RR-to-CH₄的优异性能支持了g-C₃N₄载体氮空穴中的单个Cu原子是CH₄生成的活性位点的假设。
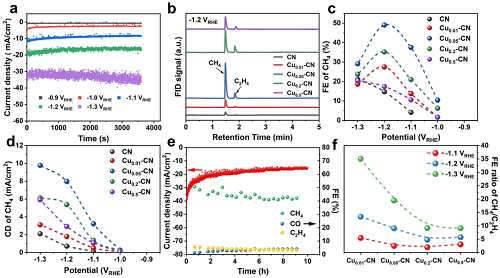
图4. CN和Cuₓ-CN催化剂的CO₂RR性能。(a) Cu₀.₀₅-CN催化剂在不同电位下的计时电流曲线;(b)气相色谱的FID信号显示Cuₓ-CN催化剂上CH₄和C₂H₄的主要气态CO₂RR产物;(c) CH₄在Cuₓ-CN催化剂上不同电位下的FE;(d)在Cuₓ-CN催化剂上CH₄在不同电位下的电流密度(CD);(e)在-1.2 VRHE下CO₂RR测试10小时期间,Cu₀.₀₅-CN催化剂的催化稳定性;(f)不同电势下Cuₓ-CN催化剂上CH₄/C₂H₄的FE比。
III CO₂RR机理和理论计算研究
为了探索CO₂RR在Cuₓ-CN催化剂上的反应机制,我们进行了原位ATR-FTIR测量,以确定CO₂RR过程中的关键中间体。如图5a所示,1268 cm⁻1和1350 cm⁻1处的峰属于*COOH中间体,它通常被认为是CO₂电化学转化为CO并进一步还原的关键中间体。值得注意的是,1130 cm⁻1和1490 cm⁻1处的信号,可以分别分配给*CH₂O和*CH₃O,随着电位从-1.0 VRHE降低到-1.2 VRHE,信号增强。因此,在Cu₀.₂-CN催化剂上形成CH₄很可能是通过*COOH、*CO、*CHO、*CH₂O和*CH₃O的质子-电子转移。为了深入了解Cuₓ-CN催化剂的原子Cu位点的形成和构效关系,我们进行了DFT计算。计算表明,g-C₃N₄ 的氮空穴中嵌入一个Cu原子的形成能为-2.11 eV,这一结果支持力g-C₃N₄中由6个氮原子组成的氮空穴是容纳单个金属原子的主要而稳定的结构。优化的结构表明,氮空穴中的Cu原子与4个N原子坐标,电荷从Cu原子转移到N原子。反应各步的自由能图表明,CO₂-to-CH₄转换的速率决定步骤(RDS)是*CO→*CHO,在Cu1-g-C₃N₄单活性位点上ΔG为1.09 eV,这比C₂H₄途径的RDS的ΔG低约0.14 eV(图5c)。因此,在g-C₃N₄限域的Cu单原子活性位点上,更有利于形成CH₄而不是其他碳氢化合物。
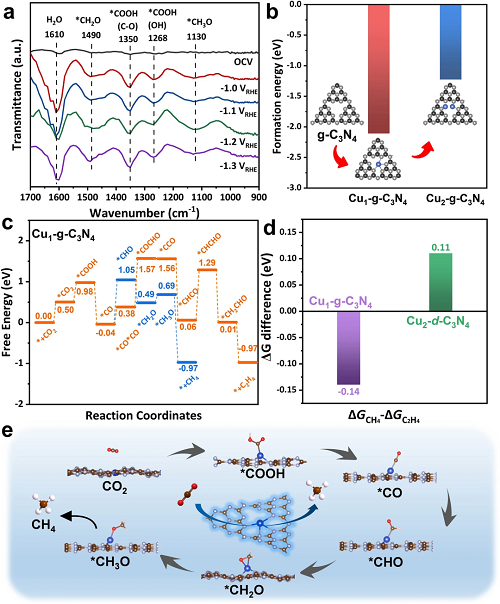
图5. 机理及理论研究。(a)Cu₀.₀₅-CN催化剂在CO₂饱和的0.1 M KHCO₃电解质中的原位ATR-FTIR光谱;(b) g-C₃N₄氮空穴中单、双Cu位点的生成能;(c)在Cu1-g-C₃N₄上CO₂到CH₄和C₂H₄的自由能图;(d)在Cu1-g-C₃N₄和Cu2-d-C₃N₄上CH₄和C₂H₄途径之间的自由能垒的差异;(e)在Cu1-g-C₃N₄上形成CH₄的反应途径。
作者简介

本文第一作者
 马元元
马元元本文通讯作者
(1)新型化学电源体系和电极材料的研发,(2)各类金属合金电催化剂的设计合成和应用研究。
▍Email:yyma@dhu.edu.cn
 杨建平
杨建平本文通讯作者
硅基、碳基、铁基、铜基、有机-无机材料界面调控用于高性能锂离子电池负极、微塑料催化降解、硝酸盐还原、二氧化碳转化与资源化。
▍Email:jianpingyang@dhu.edu.cn
关于我们
 Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 东华大学杨建平等:将CO₂高效转化为高价值甲烷的新策略,原子水平设计铜基催化剂的电化学还原CO₂(CO₂RR)
 西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化
西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化