研究背景
手性在物理、化学尤其是生物过程中起着至关重要的作用,但制备单一手性化合物需要为其“量身打造”手性催化剂,制约了合成效率。本文提出一种通用策略,利用天然的对称性破缺来实现手性检测和控制。具体地,根据手性(分子)、自旋(电子)和偏振(光子)三种自然对称性破缺之间的联系,提出利用单分子结,实现以单分子/事件分辨率精确监测与合成手性化合物的目标。相信今后通过化学家、物理学家、材料科学家和工程师的密切合作,该普适性的策略可以在精确的手性检测和不对称合成中发挥重要作用。
Chen Yang, Weilin Hu, and Xuefeng Guo*
https://doi.org/10.1007/s40820-023-01184-5
本文亮点
内容简介
手性在物理、化学尤其是生物过程中起着至关重要的作用,但制备单一手性化合物需要为其“量身打造”手性催化剂或是手性拆分材料,制约了高效合成。北京大学郭雪峰课题组提出了一种通用策略,利用天然的对称性破缺来实现手性检测和控制。手性(分子)、自旋(电子)和偏振(光子)这三种自然对称性破缺属性之间具有错综复杂的关系。厘清其内在原理并加以利用,可以实现光子偏振,电子自旋到分子手性的不对称转移,进而有望实现普适性的不对称合成。他们提出利用单分子结,以实现单分子/事件分辨率精确监测与合成手性化合物的目标。今后,相信通过化学家、物理学家、材料科学家和工程师的密切合作,该普适性的策略可以在精确的手性检测和不对称合成中发挥重要作用。
图文导读
I 不对称属性之间的联系
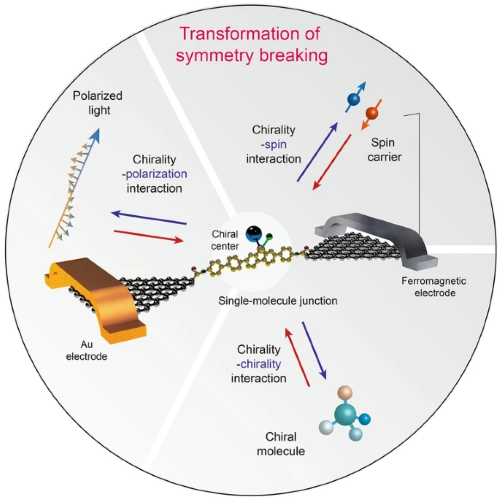
自然界中的其他对称性破缺元素,包括偏振光和自旋电子,与手性有着千丝万缕的联系,并为解决上述挑战提供了新的思路(图1)。例如,甲虫的外骨骼可以通过手性分子阵列反射偏振光。当极化电子通过手性分子传输时,也观察到自旋过滤效应(图1)。电子自旋和光子偏振之间的相互作用也很常见。例如,可以通过自旋注入实现偏振光的发射,并且可以通过偏振光的照射在半导体中产生自旋载流子。这里,主要关注是否有可能利用偏振光和自旋电子来调控手性。其中,关键是有效地构造对称性破缺属性的转移界面。在分子结中,手性分子确定的取向有助于与偏振、自旋发生有效耦合。
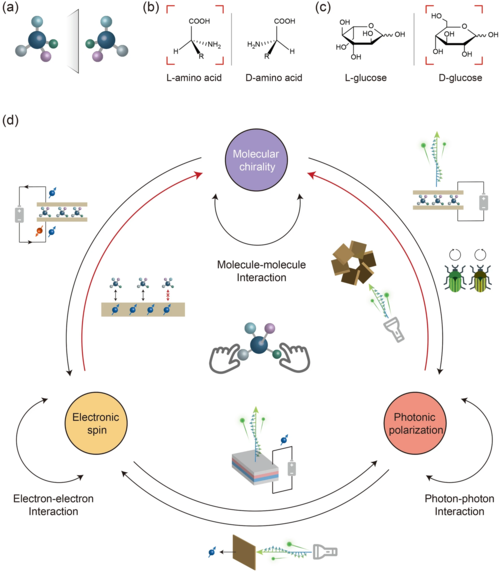
图1. 分子手性、电子自旋和光子偏振的示意图。(a) 具有镜像关系的手性分子(对映体)的示意图;(b) 手性氨基酸,其中天然的L构型被突出显示;(c) 手性葡萄糖,其中天然的D构型被突出显示;(d) 分子手性、电子自旋和光子偏振之间的关系。
II 手性-手性相互作用
基于手性-手性对称性破缺传递,是宏观监测的主流方法,也可以应用于单分子检测。例如,在纳米孔中,手性内壁与各个对映体具有不同的相互作用模式。可通过离子电流直接观察到SN2取代过程中的手性翻转(图2)。同样,在单分子结中,可利用手性的β-环糊精作为主体分子连接到石墨烯电极中(图2)。通过实时测量电导状态和停留时间可以检测相互作用过程中热力学和动力学的差异。这种单分子方法可以方便地绘制不同手性氨基酸的特征指纹图谱,有望应用于高通量检测。此外,可以通过手性伴侣分子修饰针尖,利用扫描探针显微镜测量与手性相关的高度或力来实现底物分子的手性识别(图2)。
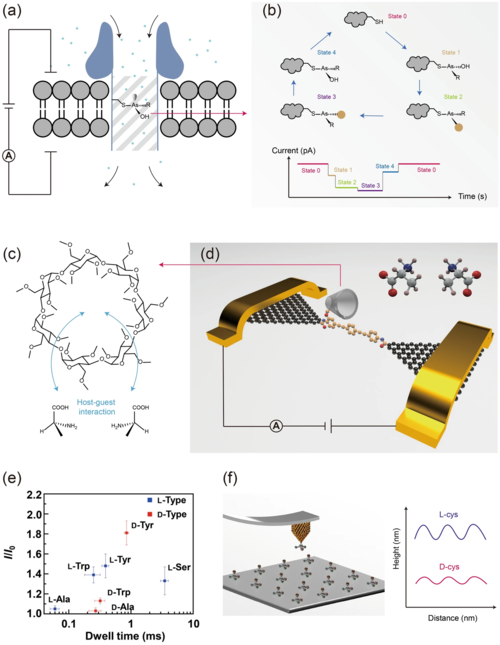
图2. 基于手性-手性相互作用的手性检测。(a) 内壁具有手性位点的纳米孔示意图;(b) 手性物种的转换序列;(c) β-环糊精和手性氨基酸之间的主客体相互作用示意图;(d) 具有β-环糊精功能中心的单分子结的示意图,用于检测手性氨基酸;(e) 氨基酸的指纹图谱;(f) 手性分子修饰针尖的原子力显微镜示意图,可检测表面上的手性分子以及绘制对映体相应的高度-距离曲线。
III 手性-偏振相互作用
自然界的电磁场对称性破缺可被应用于分子手性检测,如利用基于偏振光的圆二色谱(CD)测量分子手性,其基本原理是光与分子之间的对称性耦合。许多研究人员对不同的手性化合物甚至手性纳米材料进行了CD谱表征(图3)。Barnes等人检测了对映异构体M2和P2的圆极化(图3)。右、左圆偏振激光辐射激发M2和P2的代表性荧光强度轨迹如图3所示。CD还可以揭示了纳米材料的手性表面畸变。Govorov等人从理论上研究了纳米粒子光学手性的机理,并推测纳米粒子中的手性是来源于表面畸变导致等离子体的谐波混合 (图3)。
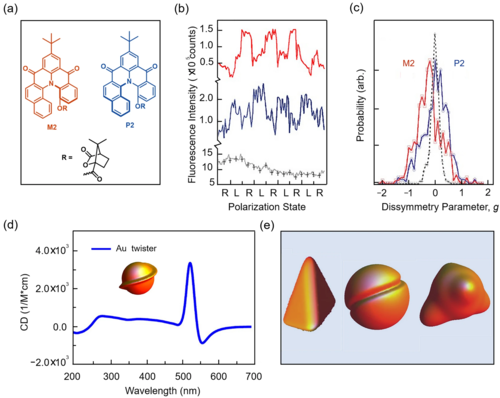
图3. 圆二色谱对手性化合物以及手性纳米材料的检测。(a) M2以及P2分子的结构;(b) 荧光强度随激发偏振光(L或R)的变化。黑线为无手性分子的对照实验;蓝线是P2分子;红线是M2分子;(c) CD谱中不对称参数的分布。蓝线是P2分子;红线是M2分子;(d) 不同手性纳米粒子的结构与CD谱;(e) 手性纳米颗粒的不同结构。
考虑到手性分子并不总是具有强的发色团,并且它们的CD信号在远紫外区可能相当弱,研究人员基于非共价相互作用、金属配位键和共价键的手性检测发展了另一种基于CD信号的手性监测方法。客体的手性基团与宿主的发色团之间的接触有助于分子识别,从而产生诱导CD信号。Biedermann和Nau报道了用葫芦脲中形成的指示染料在水中进行二元配合物的圆二色谱检测(图4),在CD光谱中显示出响应。此外,该手性检测器可用于区分不同的分析物(图4),可观察底物的范围相当广泛,包括氨基酸、多肽、蛋白质、药物分子、天然产物和小的手性有机分子(图4)。但是,这种传感器要求手性基团应该与中性芳香基团相似,以产生有效的手性转移过程。
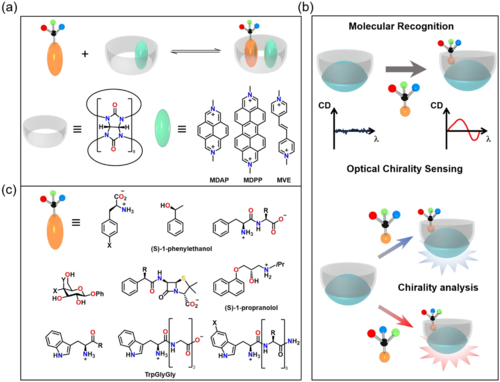
图4. 发色团辅助下的手性基团CD信号。(a) 发色团辅助的手性化合物的CD谱检测;(b)手性超分子识别示意图;(c) 底物的范围。
偏振光除了可以用来检测手性分子以外,还可以促使产生具有特定手性的化合物。圆偏振光(CPL)可以选择性地与手性分子相互作用,实现手性化合物选择性富集。此外,为了诱导自组织材料的手性,CPL已经被用于手性液晶和聚合物的合成,特别是晶体或纳米多孔材料。此外,在CPL照射和其他外部不对称刺激等条件下,非手性分子可以通过自组装构建手性纳米结构。Heinke等人提出了一种由CPL诱导的对映体选择性富集的手性MOF材料,这种MOF材料中存在着光活性的氟化偶氮苯侧基,在绿光下趋向于选择顺式构象,在紫光下趋向于选择反式构象(图5)。在顺反异构的过程中,这种材料中还表现出了不同的手性。在非偏振光下没有观察到对映体的富集,即R-和S-反式和R-和S-顺式两种异构体等量分布。然而,在CPL作用下会引起手性富集。R-反式和R-顺式对映体在右CPL作用下反应活性增强,使得S-对映体富集,反之亦然(图5)。不同类型的CPL有利于不同的手性,以产生特定手性的化合物。特别的,在偏振光下,无偶氮苯修饰的MOF薄膜上没有观察到手性对映体富集,表明偶氮苯是手性MOF成功构建的关键因素(图5)。
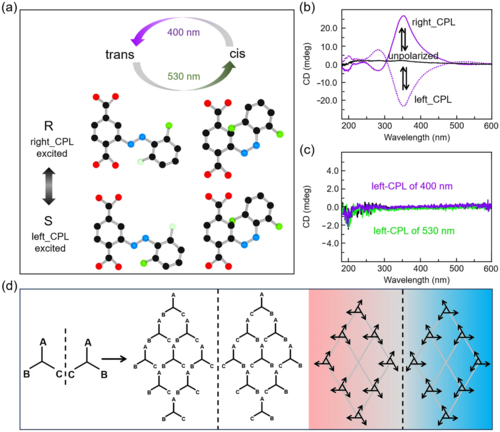
图5. CPL诱导的偶氮苯修饰手性MOF的对映选择性合成。(a) 氟化偶氮苯的光致异构化示意图;(b) 氟化偶氮苯修饰MOF在非偏振光(黑线)、右CPL(实线)和左CPL(虚线)照射下的CD光谱;(c) 不含偶氮苯侧基的MOF的CD光谱;(d) 手性化合物的组装过程示意图。
除了诱导分子的不对称合成外,CPL还被用于构建手性超分子微结构。邹等人提出了一种非手性卟啉分子TPPDA的手性自组装过程(图6)。经R-CPL和L-CPL辐照后,超分子微结构发生弯曲(图6)。他们认为CPL的角动量与卟啉环中π电子的相互作用导致了手性富集,形成的不同的手性堆叠结构将作为手性模板,在整个凝胶化过程中引导手性超分子结构的组装,使得在去除CPL辐照后产生进一步的手性扩增和转移(图6 )。
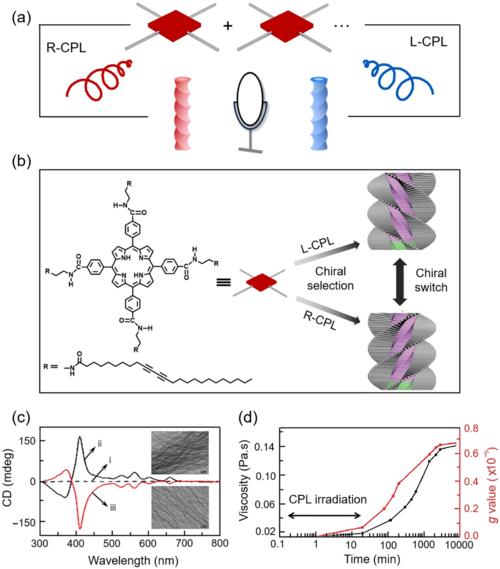
图6. CPL诱导的不对称自组装。(a) CPL诱导的超分子组装过程;(b) CPL诱导的TPPDA不对称堆叠过程;(c) 无偏振UV(i)、R-CPL(ii)和L-CPL (iii)照射20 min后的CD光谱和TEM图像;(d) CPL诱导手性自组装过程中粘度(黑色)和g因子(红色)的变化。
利用CPL进行手性检测和合成也存在一些不足。首先,该技术需要具有发色团的分子来描述局部特征。其次,偏振光检测需要纯度接近96%的样品,这给混合物检测以及原位化学反应检测带来了困难。最后,CPL对手性的具体调控尚不清楚,CPL对手性富集的影响有待进一步研究。CPL对反应的结果通常被讨论,而手性反应的微观机制很少被详细描述。单分子平台可能是研究偏振光与手性相互作用的有效方法。在单分子实验中,将单个分子连接在电极之间,通过检测其电信号和其他性质来描述分子特性。这种平台技术很大程度上排除了系综平均效应和其他杂质的影响,反映了手性分子与偏振光之间固有的相互作用,为偏振光-分子手性的检测和诱导提供了一定的指导。
IV 手性-自旋相互作用
作为自然对称性破缺的另一个特征,电子的自旋也与分子手性有着密切的关系,特别是当电子穿过分子时。1999年首次观察到手性诱导的自旋选择性效应(CISS)(图7)。其内在原理至今仍不清楚。现有模型中最直观的是手性分子(特别是螺旋分子,如DNA)提供的螺线管磁场,影响了具有不同自旋的运动电子。事实上,除了螺旋分子外,手性小分子也能表现出很强的CISS效应。另一个理论模型指出,强CISS效应主要由金属电极主导。小手性分子仅提供有限的伪磁场作为初始对称性破缺,从而诱发金属电极的轨道磁矩。CISS效应的物理起源仍然存在许多争议,这也引发了对对称性破缺起源及其关系的探索。
无论如何,CISS效应将有助于实现精确、便捷的手性检测。单分子结中电极的一侧可以采用铁磁(FM)金属实现自旋注入。在扫描探针显微镜中,具有不同螺旋结构的多肽可以无需通过手性伴侣修改尖端即可检测手性(图7)。该技术有望得到进一步推广并获得更多的应用。例如,手性的实时监测。石墨烯基单分子结具有对复杂反应环境的高耐受性和对外部刺激的兼容性,能够在反应过程中通过CISS实时原位监测手性的变化。实验发现:锚定的手性分子只能提供初始对称性破缺,从而诱导Au电极的轨道磁矩。通过金电极表面磁化强度与手性分子自旋不平衡之间的相互作用,可以观察到相同程度的CISS效应。因此,有争议的CISS效应机制被阐明,为分子手性与电子自旋之间的传递提供了新的见解。这一进展使得能够原位实时监测不对称反应过程中的手性变化,并发现关键的手性中间体、对称破缺轨迹、立体选择性分子间相互作用以及不对称反应轨迹的演化。单分子尺度。相信未来单分子结的高度集成将为实现基于自旋电子学的不对称反应的实时高通量检测铺平道路。
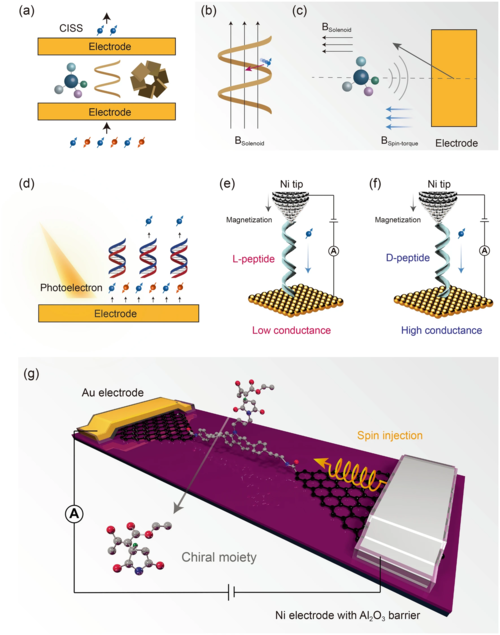
图7. CISS效应和通过自旋极化电子检测手性。(a) CISS 效应示意图;(b) CISS 效应的一种理论模型。手性分子的螺线管磁场导致对特定自旋电子的偏好;(c) CISS效应的另一个理论模型。手性分子的螺线管磁场仅提供初始对称性破缺,从而诱发电极的轨道磁矩;(d) 金属表面上的 DNA 对光电子进行自旋过滤的示意图;(e) L-多肽导致电子被向下磁化的 Ni 尖端自旋极化的过滤;(f) D-多肽导致电子的传输,电子被向下磁化的 Ni 尖端自旋极化;(g) 基于石墨烯的单分子结检测手性的示意图。
除了手性检测之外,由自旋电子控制手性也令人着迷。我们期望通过自旋特性进行的手性的分离与直接合成。对于前者,磁化的金属基底可选择性吸附手性分子(图8),包括DNA、多肽和氨基酸,然后实现拆分。分子和基质之间的相互作用会引起一般的电偶极子极化。在这种情况下,分子的手性使得电荷极化伴随着自旋极化。随着铁磁金属基底的磁化,分子自旋与基底自旋之间的交换相互作用导致对映体不同的吸附动力学,进而实现手性拆分。该技术显示出高性能的分辨率,吸附特异性高达40%以上,有望成为一种通用策略。我们相信利用电子自旋极化材料作为填料或内壁的柱色谱是未来分离手性分子的重要手段(图8)。自旋电子“催化”的直接不对称合成可能更加高效且原子经济。其实,电催化已是有机合成的常用技术。然而,如上所述,由于分子和电子的框架不固定,电子的自旋特性并未得到广泛应用。单分子结中锚定分子的严格固定可能会耦合电子的自旋特性。更具体地说,采用刚性分子桥和与电极的稳定锚定界面(图8)可能实现确定的空间方向和自旋极化电极之间的有效相互作用,进而实现直接不对称合成。单分子结的高度集成提高了大规模不对称制备的可能性。
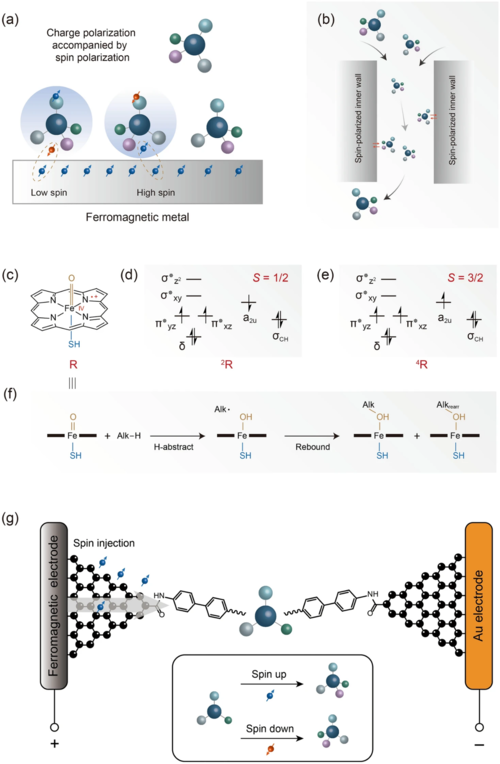
图 8. 通过自旋极化控制手性。(a) 磁化的金属表面导致对映体的选择性吸附;(b). 采用自旋极化材料拆分手性分子;(c) 铁卟啉阳离子的示意图;(d) c的低自旋态轨道占据图;(e) c的高自旋态轨道占据图;(f) c催化氧化制烯烃的反应路径;(g) 自旋极化不对称催化策略的示意图。
V 对手性起源的探索
对称性破缺之间的关系启发我们通过自旋电子和偏振光来检测和控制手性,这也促使人们思考这些对称性破缺的起源,特别是地球上的同手性。模仿自然有可能从根本上实现手性检测和控制。来自外星的圆偏振光和陨石中过量的L-氨基酸很可能造就了早期地球上生命的同手性。此外,其他理论模式也已建立,包括军官-士兵效应。也有理论表明外部能量可以使状态远离平衡并导致随机对称性破缺和手性富集。因此,不仅要考虑单分子反应,还要考虑多个分子的集体效应。手性的起源仍然是一个基本但尚未解决的核心问题。我们相信,探索中的任何进展都将为更有效地检测和控制手性提供指导。
作者简介

本文共同第一作者

本文共同第一作者

本文通讯作者
▍主要研究成果
▍Email:guoxf@pku.edu.cn
关于我们

如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 北大郭雪峰等:单分子尺度下手性化合物的精准监测、控制与合成