双氧水(H₂O₂)是一种重要的平台分子,在化工、新能源、废水处理、医药等领域有广泛的使用。目前,全世界每年大约消耗500百万吨H₂O₂,需求呈不断增长之势。蒽醌氧化法是现有的最主要的生产方法,但其能耗高,不安全,还会释放一系列有害物质。在这种压力下,清洁绿色高效的H₂O₂合成方法开始不断涌现。其中光催化/电催化合成方法因为很好兼容太阳能等可持续性新能源,同时原料为H₂O和O₂,表现出可作为终极合成方法的潜力。近十年来,有关光催化/电催化合成H₂O₂的报道不断出新,对于理论和应用的研究也不断深入,明确了基于反应理论认识上进行催化剂设计理念的重要性。
 Photocatalytic and Electrocatalytic Generation of Hydrogen Peroxide: Principles, Catalyst Design and Performance
Photocatalytic and Electrocatalytic Generation of Hydrogen Peroxide: Principles, Catalyst Design and Performance Nano-Micro Letters (2023)15: 77
https://doi.org/10.1007/s40820-023-01052-2
本文亮点
1. 系统概括了光催化和电催化双氧水合成的基础反应机理。
2. 详述了合成双氧水的光催化剂和电催化剂的设计原则,性能指标及反应机制。
3. 展望了光、电催化合成双氧水研究存在的挑战和发展机遇。
内容简介
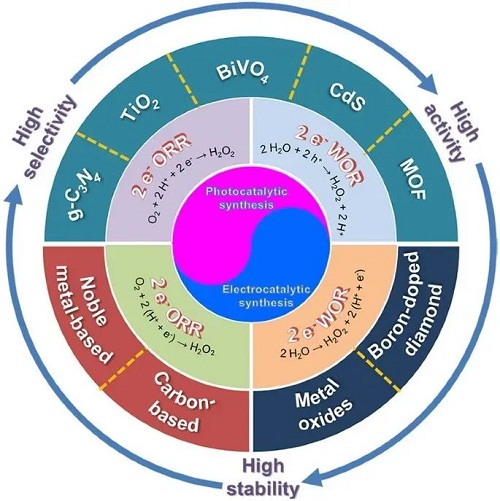
采用蒽醌法合成双氧水存在能耗高,原料污染性大,不安全等缺陷,基于太阳能等可持续能源上的光催化和电催化合成技术可从清洁的H₂O和O₂中直接合成H₂O₂,表现出巨大的发展潜力以代替主流的蒽醌法。近十年来,有关光、电催化合成H₂O₂的报道不断涌现,各种新催化材料及其反应机理先后被发现和证实。面对不断积累的研究成果,非常有必要进行系统总结以推动该领域的发展。基于此,中国科学院山西煤炭化学研究所童希立&德国锡根大学杨年俊团队近日发表了综述文章。该文章详细介绍了目前H₂O₂合成现状,表现从传统蒽醌法向光、电催化合成法过渡的趋势。分别深入总结了光催化和电催化合成H₂O₂的反应机理,并提出通过催化剂设计以实现性能提升的主要原理。在此基础上,分别归纳了光催化剂和电催化剂的设计原则、分类、性能评价标准以及相应的作用机制。最后指出面对光催化剂和电催化剂都存在反应活性,选择性以及规模还不能满足市场需要的现状,提出下一步可采取的一些有效策略来推动将来的工业化应用。
图文导读
I 光催化和电催化合成H₂O₂反应机理
光催化和电催化合成H₂O₂反应机理如图1所示。光催化过程涉及氧还原反应(ORR)或水氧化反应(WOR)。对于ORR合成而言(图1a),有两种可能的机制:间接两步单电子(O₂ → ·O₂⁻ → H₂O₂)和直接一步两电子(O₂ → H₂O₂)路径。WOR合成机制(图1b)也可以分为两种路径: 直接一步法(H₂O → H₂O₂)和间接两步法(H₂O → ·OH → H₂O₂)。此外,光催化合成H₂O₂也可能通过ORR和WOR双机制进行(图1c)。与光催化相似,电催化合成H₂O₂过程同样也能归纳为三种机制:2 e⁻ ORR (图1d),2 e⁻ WOR (图1e),ORR和WOR双路径(图1f)。
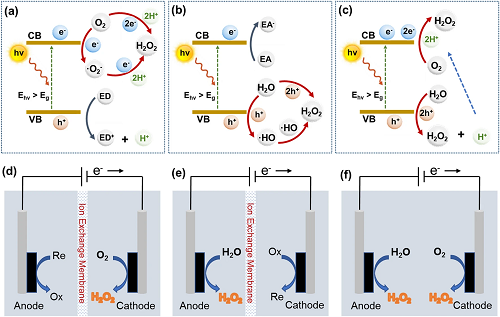
图1. (a) 2e⁻ ORR, (b) 2e⁻ WOR, (c) 双通道光催化合成H₂O₂的机理示意图;(d) 2e⁻ ORR, (e) 2e⁻ WOR, (f) 双通道电催化合成H₂O₂的机理示意图。
II 光催化H₂O₂合成
2.1 g-C₃N₄催化剂
由于g-C₃N₄的CB电位(-1.3 V)比O₂/H₂O₂(0.695 V)的还原电位更为负,因此g-C₃N₄在热力学上有潜力通过O₂还原生成H₂O₂。而且g-C₃N₄的VB电位(1.4 V)较低,能有效抑制H₂O₂的分解。然而,原始的g-C₃N₄比表面积小、捕获可见光的能力弱以及表面上O₂的化学吸附能力较低,限制了光催化合成H₂O₂性能。为此,人们提出通过制造表面缺陷、负载贵金属纳米颗粒、构建异质结复合材料、多金属氧酸盐杂化和金属/非金属元素掺杂等策略来提升性能。具体而言,缺陷具有捕获光生电子/空穴的能力,从而有效地抑制光生电子和空穴的复合;而且还能增强气体分子的吸附和活化,从而促进光催化反应进行。g-C₃N₄光催化剂的缺陷主要有两种类型:碳空位(图2a)和氮空位(图2b);负载的贵金属纳米颗粒与g-C₃N₄形成强相互作用,能提高光催化活性和选择性(图2c);具有较宽的光捕获性能的Z-型异质结有效促进光生载流子的分离,显著提高H₂O₂的产量(图2d);多金属氧酸盐(POM)与g-C₃N₄形成POM化学键,可以明显改善光催化合成H₂O₂(图2e);将金属/非金属元素掺杂到g-C₃N₄中,可以调节g-C₃N₄的带隙及载流子的传输方向,从而调节g-C₃N₄的电子、光学和其他物理性质(图2f)。
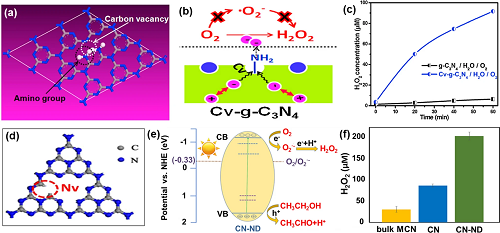
图2. (a) 含碳空位的g-C₃N₄示意图;(b) 含氮空位的g-C₃N₄的示意图;(c) 金属负载催化剂的H₂O₂浓度;(d) 异质结催化剂产生的H₂O₂浓度;(e) 3DOM g-C₃N₄-PW₁₁合成路线示意图;(f) 元素掺杂催化剂的能隙结构。
2.2 TiO₂催化剂
TiO₂的CB电位为-0.5 VNHE,足以驱动2e⁻ ORR路径合成H₂O₂。然而,纯TiO₂光催化剂产生的H₂O₂浓度仅限于微摩尔水平(< 0.2 mM)。为此,通过负载贵金属纳米颗粒、石墨烯量子点修饰以及与阴阳离子络合等手段改进TiO₂光催化剂。采用沉积-沉淀法得到的Au/TiO₂光催化剂在紫外线照射下产生的H₂O₂浓度可达10 mM,Au/TiO₂光催化剂上的反应机理解释如图3a。将硫和氮共掺杂的石墨烯量子点(GQD)与TiO₂结合(SN-GQD/TiO₂)能有明显增强的可见光吸收,H₂O₂产率可达到GQD/TiO₂的5.3倍和N-GQD/TiO₂的3.1倍(图3b)。此外,在TiO₂表面络合阳离子或阴离子也是促进光催化H₂O₂生成的有效方式。在光催化过程中,金属阳离子的内表面可以通过抑制光生载流子的表面捕获位点来促进界面电子转移;与阴离子络合的TiO₂光催化剂能抑制Ti-OOH的形成。
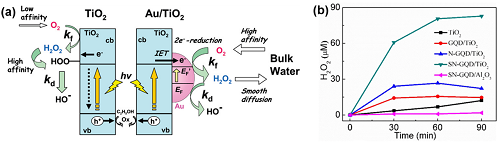
图3. (a) Au/TiO₂光催化剂上H₂O₂的生成机理;(b) 光催化H₂O₂的生成。
2.3 BiVO₄催化剂
BiVO₄具有合适的能带结构,在可见光区域具有活性。然而BiVO₄由于缺乏2e⁻ ORR的活性中心,光催化H₂O₂合成效率较低。基于此,在BiVO₄表面引入纳米Au助催化剂,由于Au和BiVO₄之间的强相互作用,促进了2e⁻ ORR路径的选择性,实现了可见光照射下生成H₂O₂(图4a)。遗憾的是,Au和BiVO₄之间形成的内置场抑制了光生电子的转移, Au表面积累了负电荷,又破坏了2e⁻ ORR路径。为此,设计了Cu@Au/BiVO₄光催化剂。Cu物种有利于光生电子从BiVO₄向Au的转移,最终促进光催化H₂O₂产生的活性增强(图4b)。
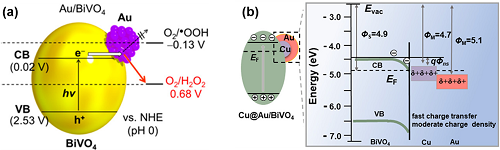
图4. (a) Au/BiVO₄光催化剂的能量图及O₂的还原电位;(b) Cu@Au/BiVO₄光催化剂的能量图。
2.4 CdS催化剂
CdS具有很好的光催化活性,但是对H₂O₂的光催化能力相对较低,这是因为CdS对反应物的吸附能力较弱,光稳定性较差。为了提高CdS在光催化H₂O₂生产中的性能,提出了以下几种改进策略。其一,将CdS和氧化石墨烯物杂化(CdS-RGO)能显著提高光催化H₂O₂合成的动力学(图5a)。其二,将贵金属络合到CdS上用于改善CdS的光催化性能。与纯CdS相比,CdS-Pt和CdS-Au纳米棒表现出更强的光催化活性以产生H₂O₂。其三,CdS/硫掺杂的碳纳米表现出优异光催化H₂O₂活性。这是由于硫掺杂碳的存在,有效地阻碍了H₂O₂的分解(图5b)。
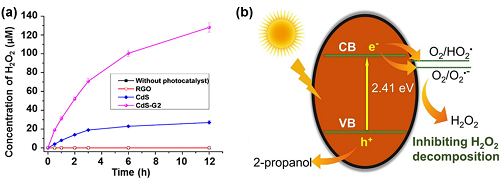
图5. (a) 不同催化剂上产生H₂O₂的浓度;(b) CdS@S掺杂碳光催化剂上H₂O₂生成的示意图。
2.5 MOF催化剂
MOF具有独特的多孔结构和强的金属-配体相互作用。与半导体类似,MOFs 在光照射下也能产生电子-空穴对。因此,它们在光催化方面显示出巨大的潜力。通过在MIL-125-NH₂上沉积NiO获得的Ni/MIL-125-NH₂,是用于光催化H₂O₂合成的第一个MOF光催化剂。H₂O₂在Ni/MIL-125-NH₂上产生的机理如图6a所示。不幸的是,得到的产物是溶解在乙腈中H₂O₂和苯甲醛,需要进一步分离和提纯。为此使用含有苯甲醇/水(BA/水)的两相体系,水相中存在原始的MIL-125-NH₂,烷基化的MIL-125-Rn位于BA相(图6b),实现了产物的分离。虽然该两相体系抑制了MOF与H₂O₂的进一步反应,但是烷基链的接枝堵塞了MIL-125-R7的孔,从而大大降低了光催化活性。居于此,开发了一种疏水性MOF,即OPA/MIL-125-NH₂,产生的H₂O₂浓度大约是MIL-125-R7光催化剂的3倍。活性的增强是由于O₂·-通过未堵塞的孔隙快速扩散所致,进而阻止了H₂O₂的分解(图6c)。OPA/Zr₁₀₀₋ₓTiₓ-MOF也表现出较高的H₂O₂产生率,有效抑制了光生电子-空穴对的复合(图6d)。此外,通过MOF和其它半导体构建异质结也能促进H₂O₂的产生(图6e)。
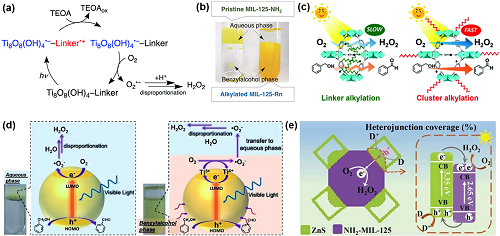
图6. (a) MIL-125-NH₂上H₂O₂生成的光催化机理;(b) 两相系统的示意图;(c) MIL-125-R7和OPA/MIL-125-NH₂的光催化过程;(d) Zr100-MOF和OPA/Zr92.5Ti7.5-MOF光催化合成H₂O₂的机理图;(e) NH₂-MIL-125@ZnS异质结光催化O₂还原为H₂O₂。
III 电催化H₂O₂合成
3.1 2e⁻ ORR 路径
3.1.1 贵金属基催化剂
根据Sabatier的原理,获得高活性需要OOH*自由基和电催化剂之间的结合能既不能太强也不能太弱。同时,还需要在转化过程中能保持O-O键的完整性。以无定形碳包覆的单金属Pt为例,无定形碳层能在铂表面产生空间位阻,诱导O₂通过“end-on”方式吸附,保证了O-O键的完整性(图7a)。对于贵金属Au而言, Au(111)表面和O₂分子之间的键太弱,使得难以形成OOH*中间体。合成Au (211)晶面则可以提高与OOH*中间体的作用力,从而有效促进2e⁻ ORR路径的活性。
相对于单金属电催化剂,合金可以进一步促进2e⁻ ORR路径的活性和选择性。与O₂弱相互作用的金属和与O₂强相互作用的金属形成合金是一个潜在的策略。例如,当纳米合金AuPd中的Pd含量达到8%时,表现出优异的H₂O₂选择性(接近95%),超过了单一金属Au和Pd催化剂(图7b)。这是因为一方面,一种金属对O₂分子表现出强烈的吸附作用,能有效地将O₂还原成OOH*;另一方面另一种技术表现弱的吸附作用,则不会破坏O-O键的完整性。
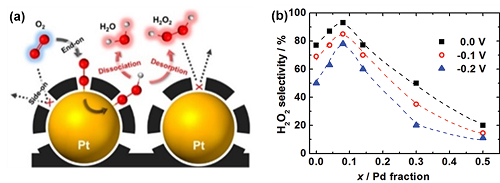
图7. (a) 碳包覆铂催化剂的ORR过程;(b) 不同Pd含量的Au1-xPdx电催化剂在0,-0.1和-0.2 V电位下的H₂O₂选择性。
3.1.2 碳基催化剂
碳材料具有独特的表面、结构性质以及低成本等优势,但本体碳材料只表现出对2e⁻ ORR的低催化活性,因此碳材料需要重构或其表面修饰。对于微孔碳(MicroC)和介孔碳(MesoC),发现在碱性条件下,它们对2e⁻ ORR均具有较高的活性和选择性,起始电位非常接近热力学平衡电位,H₂O₂的选择性甚至可以超过70% (图8a)。含氧基团的碳催化剂也很促进对2e⁻ ORR的活性和选择性。例如,氧掺杂的微孔聚苯乙烯(OMPC4,氧含量为6.52%)在碱性电解质中显示出比MPC更强的H₂O₂生成能力和更高的极限氧还原电流(图8b)。杂原子(如N,S,B)掺杂的碳材料已经被广泛用于H₂O₂的合成,例如,对介孔碳进行氮掺杂 (meso-BMP-800),可显著增强H₂O₂的选择性 (图8c)。此外,过渡金属原子分散的掺氮碳材料(M-N-C)可以稳定 *OOH、*O和*OH中间体,从而促进2e⁻ ORR路径,例如, 碳载Ni和Cu单原子催化剂倾向于在O₂还原过程中遵循2e⁻ ORR路径 (图8d)。

图8. (a) MesoC和MicroC电催化剂的H₂O₂选择性;(b) MPC和OMPC4催化剂的ORR性能;(c) 基于Koutecky-Levich曲线的转移电子数;(d) 2e⁻和4e⁻路径的活性-火山图。
3.2 2e⁻ WOR 路径
3.2.1 金属氧化物
OH*和O*的自由能是确定不同电催化剂对2e⁻ WOR选择性和活性的关键参数。同时满足ΔGO ≳ 3.5 eV和ΔGOH ≲2.4 eV的金属氧化物有利于H₂O₂的选择性生产(图9a),其中金属氧化物(如WO3,BiVO₄,MnO₂和SnO₂)有利于2e⁻ WOR的进行;金属氧化物(如IrO₂、RhO₂和PtO₂)则有利于O₂的生成。同时,归纳WOR路径产生H₂O₂的各种金属氧化物的活性可得到为火山图(图9b)。Bi₂WO₆催化剂具有较好的催化能力,可高选择性实现2e⁻ WOR进而获得H₂O₂。在一系列Bi基氧化物中,Bi₂O₃,BiVO₄和Bi₂WO₆表现出对电催化生成H₂O₂的高法拉第效率(图9c)。此外,其他金属氧化物如CaSnO₃和ZnO也被用于2e⁻ WOR H₂O₂的合成。例如,CaSnO₃纳米颗粒在3.2 VRHE时的法拉第效率为76%,高于F: SnO₂和SnO₂(图9d)。
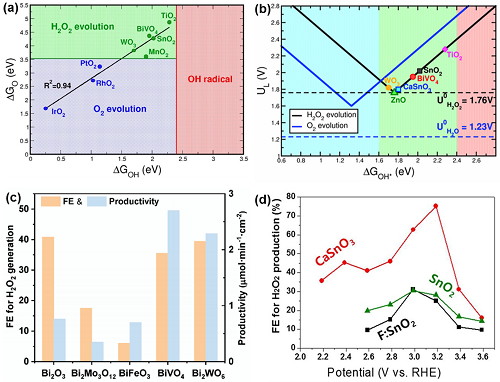
图9. (a) 用O*和OH*结合能表示的相图;(b) 2e⁻ WOR和4e⁻ WOR活性火山图;(c) Bi基氧化物的法拉第效率与产率;(d) 催化剂在不同电位下的法拉第效率。
3.2.2 BDD
掺硼金刚石(BDD)作为一种特殊的碳材料,具有极好的物理化学稳定性。2003年首次报道BDD电极上通过2e⁻ WOR合成H₂O₂。然而,FE 在3.17 VRHE时只有28%,总电流密度超过120 mA cm⁻2,表明这是一个高能耗过程。通过调整其B含量和薄膜厚度,FE可达到87%,H₂O₂产率为76.4 μmol cm⁻2 min⁻1。
IV 结论与展望
本文综述了近年来光/电催化合成H₂O₂的研究进展。在光催化合成H₂O₂的过程中,理想的催化剂需要同时满足良好的光响应(如适当的能带结构)、优异的光生载流子(即电子和空穴)分离能力以及对H₂O₂的高催化能力。然而,迄今为止报道的光催化剂合成H₂O₂活性或选择性仍然较低,未来的研究应该同时满足这三方面要求以提高合成H₂O₂效率。对于电催化合成而言,设计高活性、低成本、高耐久性的催化剂是今后发展的核心,同时需要改进反应器来以推动大规模合成H₂O₂。
作者简介

本文第一作者
金属基纳米材料的设计、制备及其在电催化领域的应用。
 童希立
童希立本文通讯作者
▍Email:tongxili@sxicc.ac.cn
 杨年俊
杨年俊本文通讯作者
先进功能材料制备、表征及电化学应用。
▍主要研究成果
▍Email:nianjun.yang@uni-siegen.de
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2021JCR影响因子为 23.655,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 中科院山西煤化所童希立&比利时哈瑟尔特大学杨年俊:光、电催化合成H₂O₂—原理,催化剂设计及性能
 西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化
西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化