研究背景
锂硫电池(LSBs)具有高理论能量密度,且所用硫资源丰富、成本低廉、环境友好,被广泛认为是下一代高性能储能器件的理想候选。然而,LSBs的实际应用受限于“穿梭效应’、电极钝化和反应动力学缓慢等问题,导致电池反应效率低、循环寿命短。传统催化机制(如吸附-催化)已取得显著效果,但仍难以从根本上突破瓶颈。近年来,三硫自由基(TRs)作为一种高电活性的关键反应中间体,因其独特的介导-催化机制成为破解上述难题的新突破口。
Breaking Boundaries: Advancing Trisulfur Radical‑Mediated Catalysis forHigh‑Performance Lithium–Sulfur Batteries
Junfeng Wu, Bohai Zhang, Zhiqi Zhao, Yuehui Hou, Yufeng Wang, Ruizheng Zhao, Hao Zhang, Jiandong Hu, Ke Yang, Bin Tang*, Zhen Zhou*
Nano-Micro Letters (2025)17: 213
https://doi.org/10.1007/s40820-025-01710-7
本文亮点
1. 本文重点强调了青金石类似物固态材料中三硫自由基的形成机制,以及高供体数溶剂及其共溶剂在稳定三硫自由基中的作用。
2. 同时探讨了三硫自由基生成过程的检测技术,这对于深入理解其行为机制及指导锂硫电池设计具有关键作用。
3. 总结了均相与非均相催化体系在促进三硫自由基生成及提升锂硫电池催化反应活性方面的策略,为其实际应用提供理论支持与技术路径。
内容简介
锂硫电池(LSBs)因其较高的理论能量密度和低成本原材料而受到广泛关注。然而,在实际应用中,LSBs仍面临多种挑战,尤其是穿梭效应、电极钝化以及反应动力学缓慢等问题。近年来,作为LSBs中的关键中间体,三硫自由基(TRs)被提出作为一种有前景的、突破传统的解决方案,能够作为“介导催化剂”提升锂硫电池的电化学性能。作为一种不同于传统LSBs中催化转化过程的体系,郑州大学周震等综述了TRs的生成、检测、促进方式及其在催化过程中的作用,尤其强调了TRs在固态青金石类似物中形成的过程,并讨论了高电子供体数溶剂及其共溶剂在稳定TRs方面的优缺点。此外,还探讨了采用均相/非均相催化剂以增加TRs数量并促进催化反应的策略。鉴于TRs在提升LSBs性能方面的巨大潜力,本文最终展望了未来TRs在LSBs中发展的方向,旨在为其深入研究提供指导。本文为电解质与催化剂的设计提供了宝贵的参考,为先进锂硫电池的实用化探索开辟了新的方向与路径。
图文导读
I S3•⁻/LiS3•自由基生成机理
1.1 固体材料中S3•⁻/LiS3•自由基稳定机理
在现代化学颜料出现之前,天然矿物青金石(Lazurite)因其迷人的蓝色被广泛用于颜料制作(图1a,1b),是古代艺术品中的重要元素,如图坦卡蒙金面具(图1c)和中国古帆船工艺品(图1d)。研究表明,这种天然蓝色颜料来源于天然钠长石结构中的三硫自由基(S3•⁻),其位于β-笼结构中,由周围的Na⁺离子稳定配位(图1e)。随着合成技术的发展,科学家可通过人工合成沸石(如LTA、FAU、EMT、LTN等)构建类似的类青金石结构(图1f),并在高温下将S3•⁻引入其中,形成具有功能性的稳定自由基材料。特别是通过引入不同的碱金属离子(如Li⁺、Na⁺、K⁺)和调控煅烧温度,能影响产物的类型和产率。此外,研究人员还开发了硅铝磷(SAPO)分子筛材料,通过引入Zn⁺并与升华硫反应,合成含S3•⁻的类青金石结构材料(如Zn@SAPO-CHA),该材料对水分子具有灵敏响应,可作为水分检测传感器。这也为其作为自由基供体催化剂在锂硫电池中应用提供了启发。
在固态锂硫电池中,尽管其能有效解决枝晶生长和多硫化物穿梭问题,但仍面临离子电导率低、固-固转化缓慢等挑战。类青金石结构中稳定的S3•⁻自由基因其良好的热稳定性、电绝缘性和潜在离子导电性,有望作为固态电解质或添加剂材料,促进SSSRR(固-固硫氧化还原反应)动力学,加快界面反应,提高循环稳定性和倍率性能。
图1. S3•⁻/LiS3•自由基稳定存在与固态材料中。a)天然青金石矿物;b)由青金石制成的群青颜料;c)图坦卡蒙法老的金面具,其上有蓝色条纹;d)一艘具有蓝色表面的中国帆船工艺品;e)装配有S3•⁻自由基的β-笼模型;f)由β-笼组成的SOD沸石结构。
1.2 溶液中S3•⁻/LiS3•自由基稳定机理
S3•⁻不仅在固态类青金石结构中稳定存在,还被发现可在高压水溶液中稳定,可能自然存在于地壳深部的高压变质环境中,并参与热液体系中金属元素的迁移及硫化物化学过程。其液相研究最早源于多硫化物溶液中的蓝色和红色现象,随后证实为硫分子在特定溶剂中通过电子转移或电化学反应形成自由基所致。研究表明,在低电子供体数(Low-DN)溶剂如DME中,Li₂S₆主要以中性形式存在,自由基生成有限(图 2a);而在高电子供体数(High-DN)溶剂如DMSO中,Li₂S₆更易解离,生成较多的S₆2⁻和S3•⁻自由基,极性溶剂的稳定作用使其反应更活跃(图 2b)。Han等人进一步提出S₆2⁻在DMSO中通过形成中间络合物(如Li₂S₆(DMSO)₄, Li₂S₆(DMSO)₈)后,经历异构化和自旋态跃迁形成LiS3•(DMSO)₂,自由基被Li⁺与DMSO配位“笼罩”,延长寿命(图 2c)。在该体系中,Li⁺展现出典型的四配位模式,与周围的氧或硫原子结合(图 2d–e),这一结构在其他溶剂体系(DMA、DME、DOL等)中也广泛存在(图 2f–h),显著影响自由基稳定性和反应路径。在多硫化物扩散调控中,需关注自由基与其溶剂化簇合物在空间尺寸上的差异,S3•⁻自由基最大尺寸约为6.4 Å,而其簇合结构可达13.1 Å(图 2i),对孔道设计提出挑战。理论计算表明,S3•⁻具有C2v对称性,A1不可约表示,展现545 cm⁻1 的拉曼特征峰和617 nm的可见光吸收峰,其n→π∗跃迁解释了其颜色来源,也为实验检测和表征提供了理论依据(图 2j)。这些研究为S3•⁻在锂硫电池中形成、稳定与调控机制的深入探索提供了理论基础和设计指导。
图2 溶液中S3•⁻/LiS3•自由基的研究示意。a)DME溶剂中多硫化物与LiS3•自由基的化学平衡图;b)Li₂S₆在DMSO中的解离路径示意图;c)S3•⁻自由基在 DMSO 中的生成机制图;d)Li₂S₆(DMSO)₄簇合物的优化溶剂化结构;e)LiS3•(DMSO)₂自由基簇合物的优化结构;f)LiS3•(DME)自由基簇合物的气相优化结构;g)LiS3•(DOL)₂ 自由基簇合物的优化结构;h)LiS3•(DOL-DME)自由基簇合物的优化结构;i)电解质体系中脱溶剂化与溶剂化的S3•⁻自由基三维分子模型与范德华表面图示;j)S3•⁻自由基的振动模式分析与电子跃迁模型图。
为了探究三硫自由基(S3•⁻)在不同溶剂中的稳定性,Lu等人借助硬酸软碱理论(HSAB)进行了解释(图 3a)。他们认为,在低电子供体数(DN)溶剂中,溶剂化能力较弱的Li⁺(硬酸)更易稳定像S₄2⁻这样的较硬碱性多硫化物;而在高DN溶剂中,Li⁺被强极性溶剂充分溶剂化,表现出较软的酸性,更有利于稳定S3•⁻、S₆2⁻和S₈2⁻这类较软碱性物种。其中,S3•⁻被认为是最软的碱,因此在高DN溶剂中含量更多。相对地,在低DN的醚类溶剂中,由于S₄2⁻稳定性强,Li₂S₆中解离产生的LiS3•自由基含量则较低。然而,HSAB理论并不能解释锂硫电池放电过程中所有现象。研究发现,LiS3•浓度在放电曲线平台(约2.0 V)开始前达到峰值,这一现象无法仅由Li₂S₆的解离解释。Park等人提出了一个补充机制,认为 LiS3•自由基还可以通过电化学反应Li + Li₂S₆ = Li₂S₃ + LiS3•生成,说明电化学条件在自由基形成过程中也发挥了关键作用,加深了我们对多硫化物转化复杂性的理解。
除了S3•⁻外,电解液中还有可能存在其他多硫化自由基Sn•⁻(n = 2–8)(图 3b)。放电过程中,正极上的环状硫(S₈)通过两电子还原形成S82⁻,并溶解于电解液中,进一步生成多种Sn•⁻自由基,它们被认为是硫还原反应的重要中间体。这些自由基可进一步获得一个电子生成闭壳层的S62⁻、S32⁻等二阴离子;也可通过对称耦合生成更长链的S102⁻、S122⁻,或通过自由基组合形成S72⁻。Prendergast等人基于实验XAS光谱与理论拟合,提出Li₂S₈可通过均裂和异裂分别生成LiS4•、LiS3•与LiS5•;Kawase 等人也通过UV–Vis光谱拟合推测可能存在LiS2•、LiS6•、LiS7•、LiS8•等自由基。然而,除了S3•⁻自由基外,尚无其他Sn•⁻自由基存在的直接实验证据,主要由于光谱信号重叠,给表征带来挑战。上述研究揭示了多硫自由基体系的复杂性,也为进一步优化锂硫电池电解液设计与反应路径调控提供了理论依据。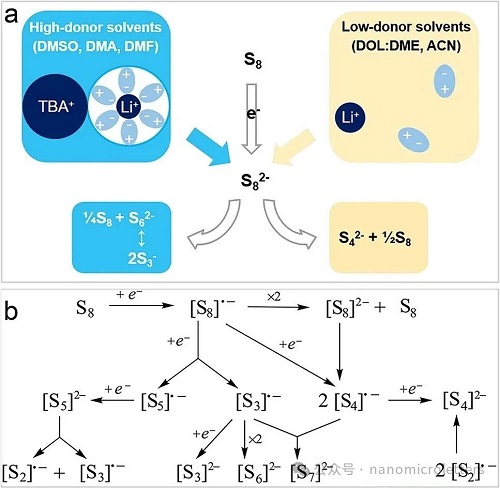
图3. S3•⁻自由基的生成机制。a)多硫化物的稳定性和物种分布如何受到其与阳离子及溶剂分子相互作用的影响汇总图b)S₈在逐步电化学还原过程中可能形成的自由基阴离子中间体示意图。
II S3•⁻/LiS3•自由基光谱检测方法
LSBs中三硫自由基S3•⁻/LiS3•⁻的生成、转化及其在反应机制中的关键作用,已通过多种先进光谱与质谱技术得到深入探究与验证。其中,磁共振类技术,如电子顺磁共振(ESR)和核磁共振(NMR)在检测未配对电子和原子核自旋环境方面具有显著优势。ESR技术利用低能微波激发顺磁中心中电子自旋态跃迁,具有较强的抗干扰能力,可在黑暗与低温条件下精确识别S3•⁻自由基的存在。实验表明,在DOL/DME 等体系中制备的多硫化物溶液普遍存在S3•⁻自由基(图 4a),且原位ESR检测到其在电化学循环过程中的含量变化明显(图 4b),说明其动态参与硫转化路径。NMR技术则可通过⁷Li核的化学位移间接捕捉自由基信号(–58 ± 10 ppm),与常规LiPSs的信号(±2 ppm)区分明显(图 4c),特别是在原位条件下,自由基信号增强,为理解其配位结构和动力学贡献提供了补充视角。
拉曼光谱则通过识别S3•⁻自由基的特征振动峰(531 cm⁻1)和其他多硫化物的峰位,分析电池运行过程中的转化路径。Liu等人在放电过程中监测到Li₂S₈、Li₂S₆、Li₂S₄等峰值的演变,并检测到微弱的LiS3•自由基信号(图 4d),表明其来源于Li₂S₆的均裂反应(Li₂S₆↔2LiSS3•)。尽管在低DN溶剂中该自由基浓度较低,但借助高灵敏拉曼信号仍可识别。进一步研究表明,当电极材料由碳布更换为金属Ni后,LiS3•信号显著增强(图 4e),归因于Ni电极表面对Li₂S₆的极性吸附效应,有利于自由基稳定与生成。这一发现为利用金属材料(如单原子催化剂)提升硫自由基参与电池反应提供新策略。
UV–Vis通过检测电子跃迁(如n→π∗)特征吸收峰(617 nm)识别S3•⁻自由基,具有优良的时间分辨能力与选择性。在DMSO中,Zou等人结合循环伏安研究发现,S₈逐步还原生成S62⁻、S42⁻、S3•⁻等中间体,S3•⁻被证明为其中最稳定且参与度最高的物种之一(图 4f)。UV–Vis技术揭示了S3•⁻与电位之间的直接关联性,但在定量分析中需注意S62⁻→2S3•⁻反应的光敏特性对吸收峰强度的干扰。
X射线吸收谱(XAS)具有穿透力强、元素选择性高、原子分辨率高等优势,可精确分析元素的价态、配位结构与局部几何环境。模拟计算显示,S3•⁻的特征XANES吸收峰出现在2468.5 eV(图 4g),明显区别于其他二价阴离子物种。进一步实验表明,在高DN溶剂(如DMA)中,自由基含量可达到总硫的25%,显著提高活性硫利用率(图 4h–i),而在低DN溶剂中其浓度低于5%,说明溶剂极性对自由基稳定性至关重要。此外,DOL在高温下与S3•⁻反应而DME不反应,提示共溶剂的温度适配性需特别考虑。
质谱(MS)技术在S3•⁻浓度极低的体系中通过稳定配体辅助增强检测灵敏度。Dou等人使用双苯甲酸(BDC)与S3•⁻自由基形成稳定络合物,进而通过UV–Vis光谱识别出612 nm吸收峰,并利用电喷雾质谱(ESI-MS)检测到[BDC–S3•⁻]络合物的特征质量数m/z = 339.4(图 4j),提供了直接实验依据,验证了配体策略在自由基识别与研究中的有效性。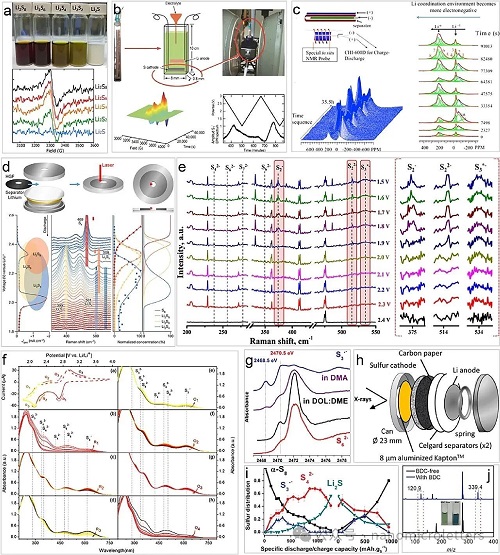
图4. 检测S3•⁻/LiS3•自由基的光谱技术。a)上图为DOL/DME溶剂中的Li₂Sₓ,下图为其对应的EPR光谱。b)用于原位测试和捕捉自由基共振信号的EPR光谱。c)原位NMR探针装置及其获得的光谱图。d)基于碳材料催化电极与DOL/DME电解液的原位拉曼测试结果。e)碳基正极与TEGDME电解液体系下LiPS还原物种演化的原位拉曼测量图。f)采用碳纸正极与DMSO基S₈正极电解液的各反应阶段的原位V–Vis光谱。g)使用液体电解液放电过程中的LSBs的K边XANES光谱,与参考材料对比图。h)用于原位XAS的在役电池装置示意图。i)循环过程中XANES光谱的线性组分拟合分析图。j)BDC缺失与BDC电解液溶液的ESI-MS质谱测试结果。
III 促进S3•⁻/LiS3•⁻自由基生成策略
3.1 高DN溶剂极其共溶剂
高DN溶剂(如DMA、DMSO、DMF)相较于传统DOL/DME电解液,具备更强的多硫化物溶解能力,尤其在低电解液/硫(E/S)比条件下,有助于实现高能量密度的锂硫电池。高DN溶剂可促进多硫化物歧化与解离反应,生成中间产物S3•⁻自由基(图 5a),该自由基可作为“氧化还原中介体”加速反应动力学、提升硫利用率。同时,Li₂S在S3•⁻介导下以3D颗粒态形式沉积,避免了电极钝化问题,维持导电性,延长循环寿命。
然而,高DN溶剂通常存在与金属锂不兼容、黏度大、离子电导率低等问题,限制了其循环稳定性和倍率性能。例如DMA、DMF等溶剂分子中的C=O或C–N强极性键易与金属锂发生副反应,生成不可逆的分解产物。在此基础上,研究发现具有独特分子结构的高DN溶剂1,3-二甲基-2-咪唑烷酮(DMI)兼具优异的极性、化学稳定性与锂金属相容性,能够有效稳定S3•⁻自由基,促进其生成(图 5b),并促使Li₂S呈颗粒态沉积(图 5c)。DMI分子结构中通过氮原子替代碳酸酯中的氧原子,降低了羰基活性,从而提升其对金属锂与多硫化物的稳定性。为进一步稳定锂负极界面并抑制穿梭效应,DMI 系统仍需配合LiNO₃添加剂使用。
除了使用SEM 观察Li₂S的3D或2D形态外,还可通过电流-时间曲线拟合判断沉积行为,常用模型包括 Armstrong、Fleischmann与Thirsk提出的AFT(n=3,3D瞬时成核)和BFT(n=2,2D瞬时成核)模型,以及Avrami方程。该方法可进一步用于探索S3•⁻自由基对硫沉积过程的调控效应。
为克服高DN溶剂带来的副反应与黏度问题,研究者提出高DN/醚类溶剂共混策略。如等体积混合高DN溶剂与DOL,可在增强多硫化物溶解与反应活性的同时,降低对金属锂的腐蚀性,兼顾动力学提升与界面稳定性(图 5d)。此类体系不仅在低E/S比下表现出更好的循环稳定性和硫利用率,同时通过调控溶剂组合可诱导 Li₂S形成颗粒状沉积,有效抑制电极钝化。
特别地,高DN溶剂四甲基脲(TMU)与DOL组成的共溶体系在软包锂硫电池中表现出优异性能(图 5e)。在DOL/TMU(1.0 M LiTFSI + 0.3 M LiNO₃)体系中,自由基显现出明显蓝色,表明高DN溶剂中S3•⁻自由基稳定存在,不受锂盐影响。TMU不仅提升多硫化物溶解性,促进S3•⁻生成,激活多路径反应机制,还能诱导Li₂S三维沉积,从而增强比容量与循环稳定性。在高E/S比下,该体系显著提升电池能量密度和寿命。但在严苛条件下电解液消耗仍限制其长循环寿命,提示在优化低E/S比设计时需重视负极电解液耗损问题。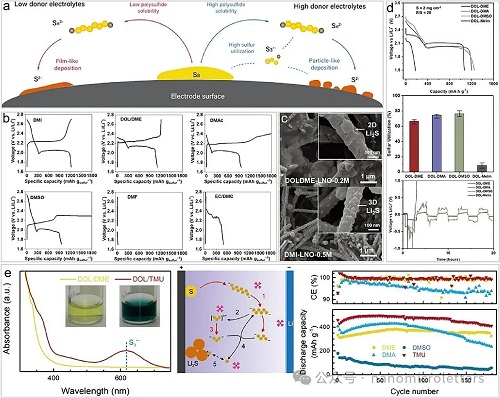
图5. 高DN溶剂与共溶剂促进S3•⁻/LiS3•自由基的生成。a)低与高给电子体电解液在LSBs中锂化过程的示意图。b)不同电解液体系下锂硫电池的充放电曲线。c)碳电极表面 Li₂S 沉积的扫描电镜(SEM)图像。d)四种混合电解液中硫正极的放电曲线、硫利用率以及锂对称电池的恒流循环性能。e)左图:Li₂S₆ 在不同溶剂中的UV–Vis光谱;中图:基于TMU的电解液的性质、机制与性能;右图:不同溶剂中纽扣电池的Li/多硫化物循环性能。
3.2 高DN溶剂添加剂
相较于以高DN溶剂为主体的电解液体系,高DN溶剂添加剂策略指在传统醚类电解液中仅引入极少量(通常 <1 vol%)高DN溶剂。这种低比例的添加,既能有效促进S3•⁻自由基的形成与稳定,又可显著改善金属锂负极的化学相容性,避免高浓度高DN溶剂所引发的副反应和锂腐蚀问题。Liang等人提出使用N-甲基吡咯烷酮(NMP)作为添加剂(<1 vol%)的策略,即为典型案例(图 6a)。该策略通过显著降低高DN溶剂与锂负极的直接接触,抑制副反应的同时,保留了高DN溶剂在促进S3•⁻自由基生成方面的独特优势。
在低浓度条件下,NMP优先与Li⁺形成稳定溶剂化层,阻止金属锂直接暴露于活性溶剂环境中,从而有效抑制腐蚀。同时,NMP可促进Li₂S的三维成核沉积行为,降低氧化物反应能垒,提高硫的转化效率和反应可逆性。这种自由基调控策略不仅减少了电解液中的副反应,还显著提升了电池的循环稳定性与容量保持率,从而延长了电池寿命,为实现含S3•⁻自由基的稳定电解液体系提供了一种高效且实用的解决方案。
然而,需注意的是,尽管高DN溶剂添加剂在低浓度下表现良好,在实际运行中,特别是低电解液/硫比条件下,仍可能对金属锂界面稳定性带来潜在挑战。因此,在该策略基础上,仍需进一步探索更高效的自由基稳定方法,如结构可控的多孔骨架材料、功能化离子液体、以及协同界面膜等手段,以全面提升LSB的能量密度与循环寿命。
图6. 高DN溶剂与阴离子添加剂促进S3•⁻/LiS3•自由基生成。a)前两图为Li₂S成核行为示意图,第三图为Li₂S₆溶液的拉曼光谱,第四图为电池中Li₂S的氧化过电位。b)左图为共盐电解液中多硫化锂(LiPSs)的UV–Vis光谱,右图为锂硫电池的充放电曲线。c)Li₂S₈溶液的UV–Vis光谱及电池容量对比图。d)左上图为不同锂盐下Li₂S₈溶液的 UV–Vis 光谱,右上图为循环后的锂负极的SEM图像及相应的充放电曲线,底图为T3Br 影响机制的示意图。e)左图为通过金属配合物平衡S3•⁻自由基的稳定性与活性的示意图,中图为不同时间的实物照片,右图为添加Al(acac)₃后Li₂S₆溶液的UV–Vis光谱。f)有机添加剂调控LSB反应机制的示意图。g)LiPSs与有机添加剂反应的模拟结果。
3.3 高DN阴离子支持电解质或电解液添加剂
除了高DN溶剂之外,引入高DN阴离子(如Br⁻、NO3⁻)的锂盐作为电解质或添加剂,即在传统醚类电解质中加入少量(通常添加剂 <0.1 M,支撑电解质 >0.5 M)高DN阴离子,可以通过其强电子给予能力有效稳定S3•⁻自由基,避免对金属锂负极的腐蚀,为优化LSBs电解液提供了更稳定、有效的新路径。相较于常见低DN阴离子(如TFSI⁻,DN = 5.4 kcal/mol),高DN阴离子如Br⁻(DN = 33.7 kcal/mol)和NO3⁻(DN = 22.2 kcal/mol)不仅能增强多硫化物的溶解度,还能抑制锂负极钝化、促进Li₂S三维沉积,提升循环稳定性。
研究表明,Chu等人在电解液中引入 0.6 M LiNO₃ 后,S3•⁻自由基浓度显著升高(图 6b),说明高DN的NO3⁻能有效稳定S3•⁻并提高硫利用率(容量超过 1200 mAh/g)。类似地,Br⁻的加入也显著提升S3•⁻含量与反应动力学(图 6c)。该策略改善了Li₂S沉积形貌,抑制极化并增强了容量保持率与循环寿命。
在此基础上,Meng等人进一步提出通过引入季铵盐(QASs)添加剂(0.1 M)并调控阳离子结构,如T3⁺、T4⁺,增强自由基生成能力(图 6d)。实验表明,T₄Br添加剂能最佳稳定S3•⁻并使溶液显现蓝色,而T₄TFSI则无此效果,显示阳离子与高DN阴离子间存在协同耦合效应。部分QAS(如T₃Br)还能通过调控Li⁺的溶剂化结构,促进SEI膜形成并改善金属锂稳定性,体现阳/阴离子协同调控S3•⁻催化机制和电极保护的双重价值。
然而,自由基的“过度稳定”会降低其电化学活性,影响有效活性物质的利用。因此,Zhao等人提出使用 0.04 M Al(acac)₃添加剂与DOL溶剂形成配位络合物,通过离子-偶极相互作用削弱Al3⁺对S3•⁻和S4•⁻的吸引,保持自由基还原活性,避免活性丧失(图 6e)。其首次提出S4•⁻的稳定机制,并鉴别出其特征UV–Vis吸收峰(676 nm)和拉曼峰(517 cm⁻1),与S3•⁻明显区分,为其他自由基种类(如S2•⁻、S5•⁻等)在LSBs中的研究与开发提供了理论参考。
此外,Zhao团队还提出使用0.1 M荧酮(fluorenone, FL)作为有机电解液添加剂来稳定S3•⁻自由基(图 6f)。FL中羰基具有电子受体能力,可捕获并稳定S3•⁻,促进Li₂S三维沉积,降低内阻并提升电池容量和循环稳定性。同时,FL可在LSBs工作电压内被还原为FL•自由基,作为电子转移中介体,加速Li₂S₄ → Li₂S的还原过程及Li₂S的氧化激活,从而改善反应动力学与整体性能。
类似地,Zhang等人使用0.05 M二苯二硒(DPDTe)作为有机添加剂促进S3•⁻生成(图 6g)。DPDTe 在放电过程中被电化学还原生成PhTe•自由基,该自由基与 Li₂S₆发生快速交换,生成电化学活性的LiS3•和中间体LiS₃TePh。后者可进一步转化为Li₂S₂、LiSTePh等中间体,并完成一个由PhTe•自由基循环催化驱动的反应路径,最终生成Li₂S。该有机硒基添加剂实现了“双自由基协同催化”机制,有效提升电池的循环寿命与倍率性能。
3.4 金属复合物催化剂
图7. 金属化合物催化剂促进S3•⁻/LiS3•自由基生成。a)上图为WO₃₋ₓ结构及其表面Li₂Sₓ转化过程的示意图,下图为含WO₃₋ₓ的硫正极随时间变化的UV–Vis光谱。b)上图为Li₂S₆吸附在ZCO量子点(ZCO-QDs)表面的优化几何构型,下图为原位UV–Vi 光谱的等高线图及基于ZCO-QDs正极的放电曲线。c)上图为电子–离子源/汇的电子传递过程示意图,下图为增强动力学的耦合反应机制图。d)左图为VS₂₋ₓ对多硫化物的吸附-催化行为示意图,右图为基于VS₂₋ₓ正极材料的原位UV–Vis光谱。e)左图为含硫空位异质结构的催化机制示意图,右图为Li₂S₆在MoS₂₋ᵧ与Co₉S₈₋ₓ晶体上的计算吸附能。f)左图为MXene材料与LiPSs相互作用机制示意图,右图为不同状态下正极材料的UV–Vis光谱。
首先,富含氧空位的金属氧化物因其增强的表面活性位点和电子结构调控能力,在催化多硫化物反应中表现出优异性能。Lin等人制备了缺氧型WO₃₋ₓ,并将其作为硫主材应用于LSBs,发现其表面可显著促进S3•⁻自由基的生成与稳定(图 7a)。此外,氧空位还能提升对多硫化物的吸附能力,抑制穿梭效应,最终实现反应动力学提升与电池高倍率稳定性增强。
不仅如此,氧化物还可通过调控金属–硫键的形成来促进自由基的生成。Liu等人研究发现ZnCo₂O₄复合氧化物可促进Li₂S₆的裂解,形成LiS3•自由基(图 7b)。其表面丰富的金属活性位点通过金属–硫配位吸附Li₂S₆,并降低其稳定性,推动其解离生成活性自由基。这些自由基进而促进固态硫的转化反应,提高了硫利用率和循环稳定性。值得注意的是,尽管ZnCo₂O₄中也存在氧空位,但该研究指出其对S3•⁻的稳定性作用不显著,主要贡献仍为增强Li₂S₄的吸附和还原效率。
Lu等人进一步提出基于分子轨道理论的电子–离子源/汇功能机制(图 7c)。在Li₂Sn-4DOL与LiSn•-2DOL之间,后者的带隙中心(BGC)更低,因此更易被还原。基于此,他们设计了Nb₂O₅/MnO₂双组分催化剂,其中放电时的LiNb₂O₅作为电子/离子源,促进LiSn• → Li₂S的还原反应;充电时MnO₂作为电子/离子汇,促进Li₂Sn → S₈的氧化过程。S3•⁻等自由基作为其中关键中间体,即使浓度较低,也在整个催化反应中扮演重要“传递桥梁”角色,说明电子结构调控策略对自由基机制的协同促进作用。
另一方面,金属硫化物催化剂同样在促进硫物种转化和提升自由基生成方面发挥关键作用。Wang等人设计了富含硫空位的VS₂₋ₓ催化剂,发现其与中间锂化相 LiyVS2-x可促进S62⁻的解离,提升S3•⁻含量(图 7d),进而加快S₈开环与多硫化物转化,有效缓解穿梭效应,增强硫利用率与反应动力学。该材料中的硫空位同时提升了结构稳定性,确保其长期催化效果。
Wei等人进一步构建Co₉S₈/MoS₂异质结,发现其硫空位并非直接使Li₂S₆裂解为LiS3•,而是先诱导其形成高反应活性的Li₂S₅中间体,再进一步转化为LiS3•(图 7e)。该机制突显出缺陷诱导的中间态调控策略在稳定S3•⁻方面的创新性与实用性,为基于缺陷工程调控自由基行为提供了理论依据。
除了金属氧化物和硫化物,新兴二维材料MXene(金属碳化物/氮化物)由于其优异的导电性、丰富的极性表面官能团和层状结构,成为LSBs催化领域的热门材料。Xiao等人合成了类花状多孔Ti₃C₂Tₓ(FLPT)MXene材料(图 7f),其表面富含羟基与氧基团,可通过Lewis酸碱相互作用有效吸附S62⁻,促进S3•⁻自由基生成并增强其稳定性。原位UV–Vis光谱检测出增强的617 nm吸收峰进一步验证该过程。FLPT的高导电性和纳米片结构加速了电子与离子迁移,使S3•⁻能迅速参与电化学反应。此外,其构建的三相界面(MXene-硫物种-电解液)进一步促进自由基富集与反应,显著提升电池容量与循环稳定性。
3.5 基碳基催化剂
图8. 碳基催化剂促进S3•⁻/LiS3•自由基生成。a)碳基电催化剂调控策略的示意图。b)使用Li₂S₆电解液的对称电池的循环伏安(CV)曲线。c)材料吸附Li₂S₆后的UV–Vis光谱图。d)碳基催化剂放电后的EPR光谱。e)不同杂原子掺杂石墨烯上LiS3•的电子密度差分图,f)其结合构型图。g)S3•⁻自由基吸附在N掺杂碳上的优化构型图。h)放电态碳/硫复合材料的ESR光谱。i)杂原子掺杂石墨烯作用下LSBs第二放电平台的反应机理示意图。j)自由基吸附吉布斯自由能与过电位关系图。
碳基催化剂具有优异的电子导电性、丰富的活性位点和可调控的表面化学性质,在锂硫电池中展现出巨大潜力。一般而言,其高比表面积和多孔结构可提供大量吸附位点与传质通道,有利于LiPSs及三硫自由基的富集与转化。其中,杂原子(如N、O、S等)掺杂是提升碳材料捕获与稳定S3•⁻自由基能力的关键策略之一。不同种类和数量的掺杂原子会影响碳材料的石墨化程度,而石墨化程度又与电子导电性正相关,例如N掺杂有利于石墨化,而O掺杂则不利。因此,合理平衡电子导通性与自由基稳定能力是碳材料设计的重要考虑。
Zhang 等人构建了N/O共掺杂的碳纳米片(UN/O-CNS)催化剂,不仅可高效锚定LiS3•自由基(图 8a),还能降低硫还原反应(SRR)和硫氧化反应(SOR)的能垒,显著提升反应动力学(图 8b)。其吸附Li₂S₆后的UV–Vis光谱表现出自由基信号强度显著降低,表明其对LiS3•有良好的固定能力(图 8c)。原位ESR测试发现,循环后的UN/O-CNS材料上出现了g值为2.035和2.053的信号(图 8d),进一步证实自由基的锚定存在。与单掺杂模型相比,N/O共掺杂诱导明显电荷重分布,三角结构活性位点的电子富集导致更强的电荷转移和更高的结合能(图 8e–f),实现了三硫自由基的稳定、穿梭效应的抑制及硫转化效率的提升。
Chen等人研究发现,氮掺杂多孔碳材料(NPC)上的吡咯N和吡啶N位点是S3•⁻的优选结合位点,其吸附能分别为–2.09 eV和–2.00 eV(图 8g)。其ESR光谱中除了常见碳自由基信号(g = 2.0023)外,还能清晰分辨出S3•⁻自由基特征信号(gy = 2.0355, gz = 2.0526)(图 8h),证实了NPC在自由基稳定中的功能,从而提升了LSBs的循环寿命与库仑效率。
为了深入理解杂原子掺杂的催化机制,Feng等人构建了一套理论模型,基于DFT计算分析杂原子掺杂对碳表面LiSy•⁻(y=1–3)及短链Li₂Sₓ(x=1–4)物种吸附行为的影响(图 8i)。研究还表明,自由基的吸附能可作为催化性能的关键描述符,用以预测反应路径、速率决定步骤与过电位(图 8j),为碳基电催化剂的理性设计提供理论支持。将DFT计算结果(如过渡态能垒、电子结构等)输入至机器学习模型中,提升高效材料筛选效率,降低实验与计算成本,为促进自由基生成、提升催化性能提供新方法。
不同于传统的自由基吸附机制,Kumar等人提出了一种创新的自由基接枝机制。他们利用富含碳自由基的活性炭布(ACC)作为硫载体,其中的碳自由基可与S3•⁻自由基发生共价接枝反应,调控硫阴极的转化路径与反应动力学(图 8k)。该机制显著改善了电池倍率性能与循环稳定性,并可推广至钠-硫电池等体系中。
IV S3•⁻/LiSS3•⁻自由基检测及增量策略的综合比较
4.1 先进检测技术
在LSBs中检测S3•⁻/LiS3•⁻自由基时,需综合考虑多项关键因素:检测精度、响应灵敏度、光稳定性、原位可观测性与原位可操作性。其中,光稳定性指的是在激光或紫外光照条件下多硫化物解离生成S3•⁻/LiS3•⁻的稳定性,越高代表光源干扰越小;原位可观测性强调在反应条件下实时观测自由基生成行为,而原位操作性进一步体现自由基行为与电化学性能的动态耦合能力。
在众多技术中,电子顺磁共振(ESR)技术表现最为突出,具有极高的精度与灵敏度,能够直接识别低浓度自由基(图 9a)。同时,其抗光干扰能力强,适合用于深入揭示S3•⁻生成与转化机制。然而,ESR仪器昂贵、操作条件苛刻、实验复杂,限制其在常规研究中的广泛使用。相比之下,UV–Vis和拉曼光谱具有良好的原位操作性与在线监测能力(图 9b、c),且操作简便、成本低廉,适合动态观测S3•⁻自由基的生成与转化行为。然而,这两种技术在光稳定性和灵敏度上略显不足,激发光源可能干扰自由基信号,因此更适合作为补充检测手段。X射线吸收谱(XAS)因其高能光子和原子分辨能力,在研究自由基的电子结构与化学环境方面具有优势(图 9d)。但其精度与灵敏度中等,且高能X光易诱发自由基或多硫化物的分解,导致光稳定性差,加之设备昂贵,操作要求较高,因此更适用于深入结构解析和搭配其他方法联用。
随着光谱技术的进步,许多新兴方法为自由基研究带来突破。如时间分辨光谱能捕捉自由基生成与转化的时间过程,揭示其反应动力学机制;双光子光谱与超快激光技术具备极高时间分辨率,可追踪极短寿命的中间自由基态;同步辐射X光谱在提升XAS分辨率与灵敏度方面表现出色,若结合原位电化学装置使用,可实现S3•⁻自由基行为与电化学性能的深入关联。
此外,多种光谱联用策略(如Raman–ESR、XAS–UV–Vis)已被应用于揭示S3•⁻自由基的生成机制与稳定过程,推动实现更全面的自由基动态分析。未来研究中,依据实验需求在精度、灵敏度与操作性之间做出权衡选择,并借助先进光谱方法与多技术协同,或将系统揭示S3•⁻/LiS3•自由基的生成–演变–稳定机制及其与LSBs性能的内在关联,为电解液设计与硫正极材料优化提供关键理论支撑。
图9. 各类光谱测试技术在检测精度、光稳定性、原位可探测性性和原位操作性方面的雷达图展示:a)ESR,b)UV–Vis,c)拉曼光谱,d)XAS。
4.2 高S3•⁻/LiS3•自由基含量催化体系的构建
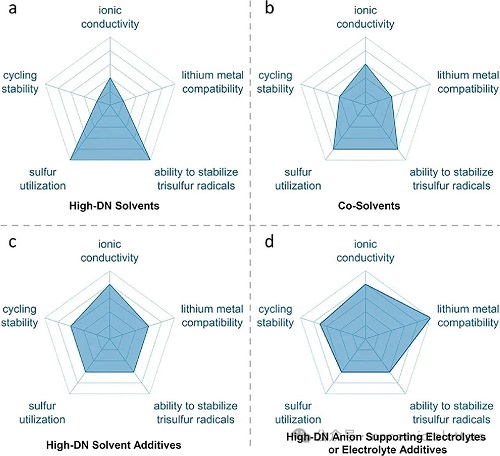
图10. 均相电催化策略在以下五个方面的雷达图展示:离子导电性、循环稳定性、硫利用率、稳定S3•⁻/LiS3•自由基的能力以及金属锂兼容性,分别对应:a)高DN溶剂,b)共溶剂,c)高DN溶剂添加剂,d)高DN阴离子支持型电解液或电解液添加剂。
在电解液策略方面(图 10),高DN溶剂因其强配位能力,能够显著促进多硫化物解离并稳定S3•⁻/LiS3•自由基(图 10a),有效提升硫利用率。然而,其对金属锂负极具有较强腐蚀性,导致循环性能受限。通过与低DN溶剂共混,以及引入如LiNO₃等功能性添加剂强化SEI,可有效缓解这一问题。共溶剂策略通过混合高DN与醚类溶剂,兼顾了离子传导性与锂金属兼容性,表现出较好的循环稳定性(图 10b)。但高DN溶剂的稀释会削弱对自由基的稳定作用。为优化该策略,可调控溶剂比例与黏度以提升多硫化物溶解与传质效率,同时选择低黏度醚类溶剂进一步改善离子迁移速率。高DN溶剂添加剂策略通过引入少量(<1 vol%)高DN溶剂,在不腐蚀金属锂的前提下,有效稳定S3•⁻自由基,实现了稳定性与效率的兼顾(图 10c),适用于综合性能要求较高的体系。结合高DN阴离子或功能锂盐,能够进一步提升整体性能。高DN阴离子型电解液或添加剂(如引入Br⁻盐)在保护金属锂、提升硫利用率与循环稳定性方面效果显著(图 10d),但对S3•⁻稳定作用较弱。优化盐–溶剂配比可提升其导电性与自由基转化效率。未来可结合机器学习与高通量计算,加速更优添加剂组合的筛选与应用。
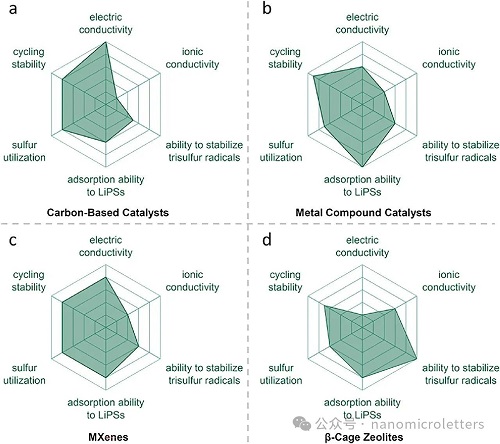
图11. 异相电催化策略在以下五个方面的雷达图展示:离子导电性、循环稳定性、硫利用率、稳定S3•⁻/LiS3•自由基的能力以及金属锂兼容性,分别对应:a)碳基催化剂,b)金属化合物催化剂,c)MXene 材料,d)β-笼型分子筛(沸石)。
在异相电催化策略方面(图 11),碳基催化剂因其高导电性与优异的循环稳定性而受到青睐(图 11a),但其对自由基与多硫化物的吸附能力有限,难以有效抑制穿梭效应。通过表面引入杂原子(如N、S、金属原子)修饰,可显著提升其化学吸附与催化能力。金属化合物催化剂具有出色的多硫化物吸附能力,可有效抑制穿梭、提高循环稳定性与硫利用率(图 11b),但其电子/离子导电性不足。通过与碳材料复合或引入氧/硫空位等结构缺陷可改善其导电性与催化活性。MXene材料兼具高电子导电性与中等离子导电性,在抑制穿梭效应与促进反应动力学方面实现良好平衡(图 11c),但其对自由基的稳定作用略弱。通过表面官能团修饰或插层金属离子等结构工程可进一步提升其自由基捕获能力。β-笼分子筛(如钠/沸石类)在离子导电性、LiPSs吸附与自由基稳定方面表现优异(图 11d),是抑制穿梭效应与促进硫转化的潜力材料。然而,其电子导电性较差,限制了其在高倍率充放电条件下的应用。可通过与导电材料复合增强其电子转移能力。
V 总结与展望
S3•⁻/LiS3•自由基作为LSBs中重要的中间体,具有促进硫转化反应、调控Li₂S沉积形貌、缓解穿梭效应和电极钝化等多重功能。理论计算方法(尤其是基于DFT的第一性原理计算、AIMD动力学与ReaxFF力场模拟)在理解其形成与转化机制中发挥着重要的作用。这些方法可从分子尺度揭示自由基在电解液与电极界面的反应路径与能垒,指导电解液与催化剂的设计,同时为光谱学特征分析提供理论支撑。
在实验层面,原位光谱表征手段(如ESR、UV–Vis、Raman与同步辐射XAS)已被广泛用于实时监测自由基的动态行为与电子结构。但因其瞬时性、低浓度与光敏性等特性,检测仍具挑战。自由基捕获剂(如亚硝酮类、吡啶鎓盐)被证明可有效增强S3•⁻稳定性并放大信号强度,结合多技术联用(如ESR–XAS、UV-Vis/Raman)将进一步提高检测精度。面向未来,时间分辨与超快激光光谱技术将捕捉其极短寿命行为,为构建自由基调控的催化体系提供突破性工具。
在材料方面,β-笼型分子筛(如青金石类材料)作为特殊自由基载体显示出独特优势。其可通过与硫反应合成,并在结构上兼具自由基稳定与多硫化物催化双重功能。通过杂原子掺杂或缺陷引入的表面工程可增强其催化吸附性能,结合石墨烯/碳纳米管等导电网络可弥补其导电性不足。同时,SAPO分子筛等人工可调结构提供了精细调控自由基生成路径与稳定性的潜力,有望拓展其在储能领域的应用边界。
尽管高DN溶剂能有效稳定S3•⁻,但其高反应性也会带来金属锂腐蚀与电解液过度消耗问题。高DN阴离子支持型电解液与有机/无机添加剂策略,通过阴阳离子协同效应实现自由基稳定与锂兼容性的兼顾,但仍需解决其长循环稳定性问题。未来可结合计算化学方法筛选低反应性高DN阴离子,并设计具有动态保护界面与人工SEI的复合或固态电解质体系。同时,原位表征将揭示自由基与锂表面在真实环境下的相互作用机理。
在催化剂方面,含缺陷的金属化合物、杂原子掺杂碳材料、MXene、新型固态催化剂等异相材料不仅具备传统吸附-催化功能,还表现出促进S3•⁻生成的潜力。但其具体机制仍待深入探索,未来需通过动态原位手段解析自由基生成/转化过程,并结合缺陷调控、导电网络构建等策略提升其稳定性与实际工况适应性(如高硫负载、低电解液用量等)。
值得关注的是,机器学习(ML)技术正快速融入LSBs材料开发流程。通过构建描述符、高通量计算与深度模型,可揭示影响S3•⁻稳定性与生成效率的关键因素,加速优质电极材料、电解质体系与催化环境的筛选。此外,潜能面学习的发展有效降低了传统计算成本,拓展了从原子尺度到器件层面的模拟空间,为全面解析自由基在电池性能中的作用机制提供强大助力。
总之,未来研究应聚焦于高DN溶剂/添加剂体系的优化设计与高效异相催化剂的开发整合,实现三硫自由基的稳定调控与反应促进。在此基础上,结合理论模拟、机器学习与先进表征方法的多维协同,将推动锂硫电池向更高能量密度、更长循环寿命与更优综合性能方向发展,并为其他金属-硫电池体系提供自由基介导的新思路与设计范式。
作者简介



▍Email:tangbin_ouc@sina.com

关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 郑州大学周震等综述:打破瓶颈–面向高性能锂硫电池的三硫自由基介导催化新进展
 青科大朱之灵/陈德宏等综述:生态纳米酶学—连接能源、环境与生态的新型催化范式
青科大朱之灵/陈德宏等综述:生态纳米酶学—连接能源、环境与生态的新型催化范式 上海大学等国际合作重磅综述:二维材料赋能神经形态计算——从原子级器件到低功耗自适应系统
上海大学等国际合作重磅综述:二维材料赋能神经形态计算——从原子级器件到低功耗自适应系统 A3-083展位|欢迎光临中国材料大会NML展位交流讨论
A3-083展位|欢迎光临中国材料大会NML展位交流讨论 韩国Cheol-Min Park等:同时适配液态/固态锂电池的多功能导电弹性复合基硅负极 原创 纳微快报 纳微快报 nanomicroletters 2026年7月13日 06:00 上海 在小说阅读器读本章 去阅读 在小说阅读器中沉浸阅读 韩国Cheol-Min Park等:同时适配液态/固态锂电池的多功能导电弹性复合基硅负极 原创 纳微快报 纳微快报 nanomicroletters 2026年7月13日 06:00 上海 在小说阅读器读本章 去阅读 在小说阅读器中沉浸阅读
韩国Cheol-Min Park等:同时适配液态/固态锂电池的多功能导电弹性复合基硅负极 原创 纳微快报 纳微快报 nanomicroletters 2026年7月13日 06:00 上海 在小说阅读器读本章 去阅读 在小说阅读器中沉浸阅读 韩国Cheol-Min Park等:同时适配液态/固态锂电池的多功能导电弹性复合基硅负极 原创 纳微快报 纳微快报 nanomicroletters 2026年7月13日 06:00 上海 在小说阅读器读本章 去阅读 在小说阅读器中沉浸阅读