A Review of CO₂ Electroreduction to Ethanol: C–C Coupling Mechanistic Insights and Catalyst Design
Fang Zhao, Bo Huang, Yingzheng Zhang, Tianxin Wei,* Jiatao Zhang, Di Zhao*
Nano-Micro Letters (2026)18: 313
https://doi.org/10.1007/s40820-026-02159-y
本文亮点
1. 机制解锁,路径革新:系统整合多路径C–C偶联机理,完整厘清乙醇生成的关键中间体与反应路径,突破传统机理认知边界。
2. 全景梳理,框架重构:全面覆盖铜基与非铜基催化剂、活性位点构效关系及原位表征技术,建立从基础到应用的一体化知识框架。
3. 策略引领,靶向设计:提出下一代高选择性乙醇电催化剂的合理设计策略,明确原子工程、串联催化与器件优化方向,为产业化提供清晰路线。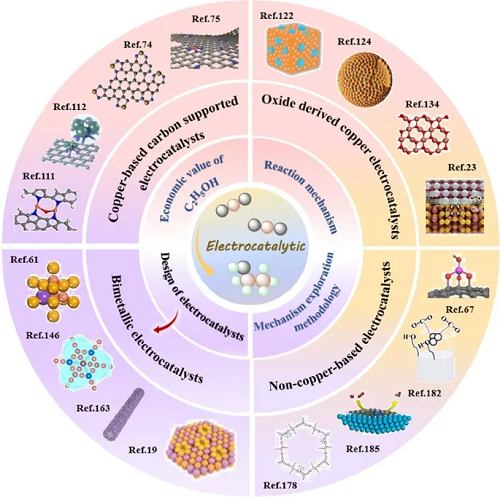
研究背景
在全球“碳中和”战略深度推进的背景下,CO₂的资源化转化成为缓解能源危机与环境压力的关键抓手。其中,CO₂电还原(eCO₂RR)制备高价值乙醇因可实现碳资源循环利用、产物能量密度高且应用场景广泛,成为科研与产业界的双重焦点。然而,该技术长期受制于三大核心瓶颈:一是反应机制复杂,涉及多电子转移与C–C耦合过程,路径繁琐,产物选择性难以精准调控,副反应干扰严重;二是催化剂稳定性不足,铜基等主流催化体系易发生结构重构,活性位点快速失活,难以满足长循环运行需求;三是实用化难度大,高载量、低能耗、高稳定性的催化体系构建困难,制约了从实验室到工业化的落地。
内容简介
北京理工大学张加涛课题组系统解析反应机制与催化动态变化,提出高效调控策略,取得突破性进展,为推动CO₂电还原技术的产业化应用提供了核心支撑。本文聚焦CO₂电还原(eCO₂RR)制乙醇领域,系统梳理了该领域的研究进展、核心成果及发展趋势。文章重点汇总了CO₂电还原制乙醇的四大核心反应路径,系统分析了*CO二聚化、两个*CH₂物种耦合、CO插入*CH₂三条成熟路径的关键速率决定步与调控方式,同时归纳了非铜基催化剂(锡基)上*CO(OH)与*CHO耦合的全新路径研究进展,打破传统单一催化研究的局限。针对催化剂稳定性这一领域共性问题,综述了催化剂动态重构调控策略的研究现状,总结了铜基催化剂从单原子到纳米颗粒、铜簇的结构转变规律,以及CeO₂添加、硫空位引入等改性手段的应用效果。此外,整合了原位红外光谱、原位拉曼光谱、原位同步辐射及DFT计算四大表征技术在该领域的应用,从原子尺度总结反应全过程与催化剂变化的研究成果,并梳理了铜基碳负载、铜基合金等高效催化剂体系的研究进展,为该领域后续研究方向及工业化应用提供了全面的参考与指导。
图文导读
I 历程“时间轴”:CO₂电还原制乙醇的研究演进全景
如图1所示,这条时间轴完整展现了CO₂电催化还原制备乙醇的发展脉络。从最初明确乙醇生成依赖C–C偶联与12电子转移过程,到确立铜基催化剂的核心地位;再到通过晶面调控、价态优化、缺陷工程逐步提升选择性;最后发展到双金属协同、单原子催化、非铜基催化等突破性体系,实现高选择性、高电流密度与长稳定运行,清晰勾勒出该领域从基础机理走向工业化应用的发展趋势。
图1. 通过eCO₂RR的乙醇生产时间线。
II 框架“总览图”:CO₂电还原制乙醇综述核心内容全景
图2系统呈现了本篇综述的整体逻辑框架与核心研究维度。围绕CO₂电催化还原制乙醇这一主题,依次从乙醇经济价值分析、多路径反应机理与原位表征方法、铜基与非铜基催化剂活性位点及构效关系、电化学性能指标对比,到下一代高性能催化剂理性设计策略五大模块展开,全面覆盖基础机理、材料设计、性能优化与应用导向,构建起该领域从理论到实践的完整知识体系,为后续研究提供清晰的思路指引。
图2. 本文所讨论的主题概述。
III 价值“风向标”:CO₂电还原产物的热力学与经济性图谱
图3直观揭示了CO₂电还原各类产物的热力学特性与经济可行性。上方展示乙醇生成所需的多电子转移过程与标准还原电位,突出其12电子转移的复杂反应特征;下方对比不同产物的市场价格与单位能耗,清晰表明乙醇作为C₂液体产物,兼具高体积能量密度、大市场规模与优异的成本竞争力,是CO₂电还原中最具工业化价值的多碳目标产物,为研究方向提供了明确的经济决策依据。
图3. 二氧化碳电解产物的热力学与经济概况。
IV 路径“导航图”:CO₂电还原制乙醇的多元反应路径
图4展示了CO₂电催化还原生成乙醇的多条可能反应路径。核心围绕C–C偶联这一关键步骤,主要包括*CO二聚、*CO与*COH/*CHO不对称偶联、*CH₂偶联、*CO插入*CH₂,以及非铜基催化剂上CHO–*CO (OH)偶联等典型通道。不同中间体经耦合、质子耦合电子转移与逐步加氢,最终定向生成乙醇,清晰揭示了产物选择性与中间体演化、偶联方式的内在关联,为机理理解和催化剂设计提供直观路径依据。 
图4. eCO₂RR转化为C₂H₅OH的几种可能反应途径。
V 动态“演变图”:Cu基催化剂电还原过程中的原位结构重构
图5揭示了Cu基电催化剂在CO₂电还原反应中的动态重构机制。多种Cu基材料在反应电位下,会从单原子、Cuⁿ⁺配位态等初始结构,原位演变为Cu纳米团簇、Cu⁰/Cu⁺混合价态等真实活性位点;同时,掺杂、缺陷、界面调控等策略可稳定 Cu⁺活性相,促进*CO中间体吸附与C–C偶联。整套图示清晰展现了催化剂“结构—价态—活性”的动态关联,为精准设计高选择性乙醇电催化剂提供关键机理支撑。
图5. a Cu0.5NC在CO₂还原反应过程中可逆金属位点重构机制的示意图。b 基于原位测量提出的假设反应机制。c 采用Cu₂S1-x电催化剂通过eCO₂RR生产乙醇的机理示意图。d CeO₂/CuS重组过程的示意图。e 采用Cu₂(OH)₂F通过CO₂还原制备乙醇的机理示意图。f C₂H₅OH处理过程中电催化剂结构演变的示意图。
VI 原位“探测仪”:机理研究的先进表征技术合集
图6汇集了红外、拉曼、同步辐射、DFT计算四大核心原位表征手段,直观呈现CO₂电还原制乙醇的中间体演化与活性位点本质。原位红外实时捕捉*CO、*COCOH等关键中间体的吸附与转化,原位拉曼追踪Cu⁺物种与催化剂晶相动态变化,同步辐射精准解析Cu原子配位与价态演变,DFT计算则定量揭示C–C偶联与加氢路径的能垒差异。四类技术交叉验证,从分子、原子层面完整解码反应机理,为催化剂理性设计提供坚实的原位证据与理论支持。
图6.在-0.4 V至-1.2 V(vs RHE)的电位窗口内,a Cu-I-Cu和b Cu-Cl-Cu双位点的原位ATR-SEIRAS光谱。c Cu和d Cu=N电催化剂在-0.1至-1.2 V(vs RHE)电位窗口内的原位红外光谱。e Cu-I-Cu和f Cu-Cl-Cu表面吸附中间体的原位拉曼光谱。在532 nm激光激发波长下,g CuAl₂O₄/CuO 和 h CuO 在OCP及不同施加电位下进行eCO₂RR时的原位拉曼光谱。i Cu-NBA在eCO₂RR过程中不同电位下的Cu K 边原位XANES光谱。j Cu-NBA在eCO₂RR过程中不同电位下的对应傅里叶变换 FT(k³w(k))。k Cu/C-0.4电催化剂经16小时恒电流法测量后的k²加权R空间EXAFS数据及其傅里叶变换,Cu(AcAc)₂作为参考样品。l 预处理Cu/C-0.4、在-0.7 V vs RHE和-1.0 V vs RHE电位下的Cu/C-0.4,以及后处理Cu/C-0.4的原位Cu k边XANES光谱。乙醇生成关键反应中 m 初始态、n 过渡态和 o 终态的俯视图,以及乙烯生成关键反应中 p 初始态、q 过渡态和 (r) 终态的俯视图。红色、白色、灰色和橙色球分别代表氧、氢、碳和铜,而粉红色球代表Cu上的氢。图 t 显示了*COH和*OCCOH形成的自由能差,以及*CO、*COH和*OC−COH在CuBr-DDT上的优化吸附结构。u *C−CH和*HC−CHOH形成的自由能差,以及*HC−COH、*C−CH和*HC−CHOH在CuBr-DDT上的优化吸附结构。
VII 活性“分布图”:金属电催化剂的CO₂还原产物分类规律
图7基于中间体吸附能差异,对不同金属在CO₂电还原反应中的产物分布进行了系统性归类。金属对*H与*CO中间体的吸附强度直接决定催化选择性:Cu 因对*CO吸附适中、对*H吸附较弱,成为唯一能高效实现C–C偶联生成乙醇等 C₂₊产物的金属;其他金属则倾向生成CO、甲酸或H₂等单一产物。这一规律直观阐明了Cu基催化剂在乙醇合成中不可替代的优势,为催化剂体系筛选提供了核心理论依据。
图7. a eCO₂RR金属电催化剂主要产物的分类。b 二氧化碳还原金属的分类。
VIII 原子级“活性中心”:铜基碳载催化剂的结构与性能调控
图8与图9共同揭示了氮掺杂碳负载铜基电催化剂的原子级结构、原位演化与乙醇合成性能。从CuN4单原子、CuOCu–N4双核位点,到Cu团簇与Cu–N–G界面结构,研究通过精准配位环境调控,实现活性位点从单原子向团簇的动态重构,并借助氮掺杂碳基底强化电子传递与*CO中间体吸附。多种催化剂均展现出低过电位、高乙醇法拉第效率与优异稳定性,直观验证了原子级分散、双位点协同、原位结构演变对提升C–C偶联效率与乙醇选择性的关键作用,为高性能铜基碳载催化剂设计提供明确范式。
图8. a Cu0.5NC的示意图。b 在0.1 M CsHCO₃水溶液中,以2.5 mLmin⁻¹的CO₂流速下,Cu0.5NC在不同施加电位下的法拉第效率。c Cu0.5NC在无外加电位(蓝线)、-1.2 V vs. RHE电解过程中(红线)、电解后无外加电位(绿线)以及-1.2 V vs. RHE电解后样品暴露于空气中10小时(橙线)时的实验EXAFS光谱傅里叶变换图。d Cu−1/hNCNC的TEM图像。e Cu−1/hNCNC在不同电位下的FE、j及产物分布。数据为三次重复测量的平均值。误差条标注于乙醇和总产物数据。f 乙醇(路径1和路径2)、甲酸及乙酸的eCO₂RR自由能图。
图9. a 基于原位XAS和准原位XPS分析提出的催化活性Cun-CuN₃簇可逆形成方案。b Cu/N0.14C的TEM图像。c Cu/N0.14C各产物的FE。d 用于电催化CO₂还原的Cu@N掺杂石墨烯电催化剂的制备示意图。e Cu-N-G电催化剂在选定电位下的产物法拉第效率。f Cu-SACs-N-CQDs电催化剂的整体合成过程。g Cu-SACs-1N-CQDs的原位XAS表征。h Cu-SACs-1N-CQDs在不同电位下的法拉第效率。
IX 界面“调控师”:氧化物衍生铜与复合结构的乙醇选择性强化机制
接下来系统展示了氧化物衍生铜基催化剂及异质结构在CO₂电还原制乙醇中的构效关系(图10和图11)。通过Ag、Al、Ti等元素掺杂以及界面、核壳结构设计,有效稳定Cu⁺活性位点、调控*CO吸附构型并促进不对称C–C偶联;碳层与氧化物载体进一步抑制活性相重构、优化中间体加氢路径。两类结构均显著提升乙醇法拉第效率与分电流密度,清晰揭示了价态调控、界面工程、吸附组态优化对定向生成乙醇的关键作用,为高选择性催化体系构建提供了可靠策略。
图10. a dCu₂O/Ag₂.3%表面增强乙醇生成的示意图。b 在选定电流密度下,dCu₂O、dCu₂O/Au2.3%和dCu₂O/Ag₂.3%体系中C₂₊产物的FE值。c se-Cu₂O/Ag的合成示意图。d se-Cu₂O/Ag在脉冲电解条件下eCO₂RR过程的示意图。e 在CO₂饱和的0.1M KHCO₃溶液中,脉冲电解条件下不同施加电位下C₂₊产物的FE(左轴)及FEEtOH/FEC₂H4(右轴)(Ea = +0.4 V vs RHE,ta = tc = 0.5 s)。f eCO₂RR的乙醇和乙烯反应路径示意图。g Al−Cu/Cu₂O在不同电位下测得的原位ATR-FTIRS光谱。h Al−Cu/Cu₂O在不同电位下各种产物的FE分布图。
图11. a Cu₂O-TiO₂合成的示意图。b 在0.5 M KHCO₃电解液中进行一小时电解时,不同施加电位下Cu₂O-TiO₂的CO、C₂H₅OH和HCOOH法拉第效率。c Cu₂O与TiO₂之间电子相互作用的示意图。d Cu₂O、Cu₂O-TiO₂和TiO₂的CO-TPD光谱。e Cu₂O 和Cu₂O-TiO₂上CObridge与COatop的电位依赖性比值及相应的乙醇FE。f CuOx@C 和CuOx电催化剂的制备示意图。g CuOx@C在不同电流密度下进行eCO₂RR时的原位拉曼光谱。h CuOx@C和CuOx电催化剂上乙醇的法拉第效率。
X 双金属“协同器”:原子掺杂与异相界面助力高效乙醇合成
然后又集中展现了双金属调控、原子掺杂与界面工程CO₂电还原制乙醇的显著提升作用(图12与图13)。通过构建CuSn、CuMg、CuAg等异核双位点,以及V、K等杂原子掺杂Cu₂Se体系,可精准调控电子分布、稳定Cu⁺活性相、促进*CO与*CH₂等中间体的不对称C–C偶联,并有效稳定乙醇关键加氢中间体。这些策略大幅提高乙醇法拉第效率、分电流密度与稳定性,清晰揭示了双金属协同、电子调控与缺陷工程在定向合成乙醇中的核心价值,为高性能催化电极提供了高效设计思路。
图12. a CuSn-HAB的合成过程示意图。b 以CuSn-HAB为电催化剂时,不同电位下的FEEtOH值。c 在1 M KOH电解液中,CuSn-HAB作为电催化剂进行eCO₂RR反应时,-0.57 V vs RHE下的随时间变化的原位ATR-FTIR光谱。d 利用铜、镁和碳粉末合成(111)晶面取向纳米Cu₂Mg金属间化合物的示意图。e Cu₂Mg(111)电催化剂在CO₂还原过程中的FE。f Cu₂Mg(111)表面上*CHCOH中间体加氢生成乙醇和乙烯的自由能图。g 掺银铜表面上CO₂电还原为乙醇的可能反应机理。h CuAg-0.75%表面上不同施加电位下CO₂还原产物的法拉第效率。所制备的CuAg-0.75%样品的形态表征:i SEM和j TEM图像。
图13. a V掺杂Cu₂Se纳米管的生长过程示意图。b Cu1.22V0.19Se生成的各种产物的FE。c Cu1.22V0.19Se上eCO₂RR反应的时变原位DRIFTS光谱。d K掺杂Cu₂Se上eCO₂RR制备乙醇的催化机理示意图。e 不同电位下K11.2%-Cu₂Se的乙醇、CO和H₂的FE。f K11.2%-Cu₂Se的扫描电子显微镜图像。
XI 非铜“新赛道”:多元非铜基催化剂的高选择性乙醇转化机制
图14. a SnS₂@Sn₁O3G的制备流程示意图。b 带误差条的电位-电流(FE)曲线。c 示意图展示了SnS₂@Sn₁O3G上CO₂还原为乙醇过程中的级联反应。Ag0.015In0.985Se0.734的d SEM图像和e TEM 图像。f 不同电池电压下 Ag0.015In0.985Se0.734上产物的FE和乙醇的EEs。g Ag0.015In0.985Se0.734在- 0.6V下的时变原位DRIFTS光谱。h CO₂⁺金属化TAPA-OPE共价有机框架(COF)的合成。i 不同电位下eCO₂RR反应期间的产物分布图。j 电解前、电解期间及电解后Co-TAPA-OPE COF的Co K边XANES光谱。插图显示了放大的Co K边XANES光谱。
图15. a PAF-PA5-Ag-0.8和PAF-PA5-Ag-1.9的制备及其对CO₂吸附的示意图。b PAF-PA5-Ag-0.8上的产物分布。c PAF-PA5-Ag-0.8、PAF-PA5-Ag-1.9、AgNPs和PAF-PA5的CO-TPD谱图。阴影区域内的峰值源于高温下PAF-PA5-OH的分解。d Ag纳米线硫化生成Ag/Ag₂S纳米线的示意图。e 在Ag纳米线和Ag/Ag₂S纳米线上进行CO₂电还原时,不同产物的FE图像。f 在饱和CO₂的0.5 M KHCO₃溶液中,不同电位下Ag/Ag₂S纳米线电还原CO₂的SERS光谱。Ni@NCNT-700的g SEM图像及h TEM图像。i Ni@NCNT-700的FE曲线。j Ni@NCNT-700在-0.6至-1.2 V电位范围内的原位拉曼光谱。
最后,全面总结了非铜基电催化体系在CO₂制乙醇中的突破性进展(图14与图15)。锡基、银基、铟基、钴基COF、镍基@氮碳等非铜催化剂,通过双活性中心、原子掺杂、异质结、限域结构等设计,开辟出与铜基不同的C–C偶联新路径,可高效吸附并稳定* CHO、*CO (OH)等关键中间体,显著抑制副反应并实现超高乙醇选择性。系列结果打破了铜基材料的主导格局,证明非铜体系同样可实现高选择性、高稳定性乙醇制备,为CO₂电还原制乙醇提供了全新材料体系与催化机制。
XII 元素“作用谱”:可促进乙醇生成的C–C偶联位点元素汇总
图16对已报道的、能够提供有效二聚位点以推动乙醇生成的元素进行了直观分类与归纳。图示清晰展现了除核心Cu元素外,Sn、Ag、In、Co、Ni、Mg、V、K等多种非铜元素,均可通过构建双活性位点、异质界面、电子调控与缺陷结构等方式,参与并促进CO₂电还原过程中的C–C键形成。该图全面呈现了乙醇合成催化剂的元素选择范围,打破了传统铜基体系的局限,为开发新型高效、低成本、高选择性的电催化剂提供了丰富的元素库与多元化设计思路。
图16. 已报道的可作为二聚化位点、从而促进乙醇生产的元素。
XIII 未来“路线图”:高效乙醇电合成催化剂的理性设计策略
结论与展望部分凝练出四大关键方向:精准调控铜基催化剂的晶面、尺寸与表面形貌;利用缺陷工程与杂原子掺杂强化C–C偶联;通过表面官能团修饰与分子工程稳定关键中间体;并系统性优化电解池微环境与器件结构,重点发展流动池与MEA体系以提升工业适用性,全方位推动CO₂电还原制乙醇走向高选择性、高稳定性与规模化应用。
XIV 总结
本综述系统梳理了CO₂电催化还原制乙醇的最新研究进展,从经济可行性、反应机理、催化剂设计到工程化策略进行了全方位总结。乙醇作为高能量密度、易储运的液体C₂产物,在技术经济性与市场规模上均具备显著优势,是CO₂电还原最具产业化前景的目标产物之一。当前研究已明晰乙醇生成依赖C–C偶联与12电子转移过程,*CO、*CHO、*CH₂等中间体的吸附与耦合行为直接决定反应选择性。
铜基催化剂仍是实现高效乙醇合成的核心体系,通过原子级分散、价态调控、双金属协同、界面工程与缺陷工程,可有效稳定Cu⁺活性位点、优化中间体吸附、促进不对称C–C偶联,大幅提升乙醇法拉第效率与电流密度。与此同时,非铜基催化体系取得重要突破,基于锡、银、铟、钴、镍等活性中心,通过双位点耦合、限域效应、异质结调控等方式,开辟了区别于铜基的全新反应路径,实现了高选择性乙醇制备,拓展了材料设计边界。
原位表征技术与理论计算为机理研究提供了关键支撑,红外、拉曼、同步辐射等手段实时追踪中间体演化与催化剂动态重构,DFT计算定量揭示能垒与选择性来源。面向工业化应用,未来需进一步提升催化剂稳定性、抑制析氢副反应、提高大电流密度下的选择性,并结合流动池、膜电极等器件优化反应微环境。本综述为下一代高选择性、高稳定性、高活性乙醇电合成催化剂的理性设计提供了清晰方向,助力CO₂资源化利用迈向实用化。
作者简介

关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2024 JCR IF=36.3,学科排名Q1区前2%,中国科学院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 北理工张加涛团队赵娣和韦天新最新综述:CO₂高效转化制乙醇:催化剂设计与机理研究新突破