研究背景
基于绿色植物仿生模拟,人工光合作用的研究为应对全球环境恶化和可再生能源短缺等问题提供了新的解决思路。在众多光催化中,氧卤铋材料因其晶体可调性和对组成结构高度依赖的光催化特性而展现潜力。然而,氧卤铋中有限的活性位点和较低的电荷分离效率限制了其进一步应用。尽管可以通过构建异质结构加强电荷分离并引入新的外源活性位点,但其位点状态和数量难以精确调控,且电荷传递过程仍受接触界面影响。研究发现,金属卟啉基单原子层材料的二维超薄结构可以强化电子传递的各向异性,同时均一的周期性结构能够使得所有金属催化位点高度统一。若能将其引入卤氧铋光催化剂表面将有望实现催化剂高密度活性位点的构建,但如何实现二者的有效复合并调控光生电子在材料间的定向迁移行为仍是巨大挑战。
Strain-Induced Surface Interface Dual Polarization Constructs PML-Cu/Bi₁₂O₁₇Br₂ High-Density Active Sites for CO₂ Photoreduction
Yi Zhang, Fangyu Guo, Jun Di*, Keke Wang, Molly Meng-Jung Li, Jiayu Dai*, Yuanbin She*, Jiexiang Xia*, Huaming Li
Nano-Micro Letters (2024)16: 90
https://doi.org/10.1007/s40820-023-01309-w
本文亮点
1. 应力诱导Bi₁₂O₁₇Br₂与铜卟啉基单原子层(PML-Cu)强耦合,构建具有Bi-O键合界面的PML-Cu/BOB(PBOB)。
2. PBOB的表面界面双极化作用能够强化内建电场,促进电子定向转移。
3. PBOB中PML-Cu的引入为其提供了高密度分散的活性Cu位点,显著提升材料的光催化CO₂还原能力。
内容简介
光催化剂活性位点不足及界面电荷转移缓慢限制了光催化CO₂还原的效率。如何同时应对上述关键问题是一项艰巨而富有挑战性的任务。浙江工业大学佘远斌、国防科技大学戴佳钰等提出了通过应力诱导策略在铜卟啉基单原子层(PML-Cu)和Bi₁₂O₁₇Br₂(BOB)中构建Bi-O键合界面,从而引发PML-Cu/BOB(PBOB)表面界面双极化。在这种多步极化过程中,界面之间形成的内建电场将诱导电子从BOB的导带(CB)转移到PML-Cu中,并抑制其反向电子迁移;PML-Cu的表面极化进一步促进了电子在铜原子中的聚集。同时,PML-Cu的引入赋予了PBOB表面高密度分散的Cu活性位点,显著促进了材料对CO₂的吸附、活化以及CO的解吸。PBOB的光催化CO₂还原性能得到大幅提升,分别是BOB和PML-Cu的7.83倍和20.01倍。这项工作为催化剂的多步极化调控和活性位点设计提供了见解。
图文导读
I PBOB材料的设计原理及其结构特征
图1a所示为PBOB的制备策略,以Cu-TCPP为基元构建了铜卟啉基单原子层(PML-Cu)。材料以CuO₄和CuN₄分别作为卟啉环内的金属节点和配体位点,通过自下而上的交联反应生长。同时,通过应变工程制备了超薄BOB纳米管结构,其弯曲的管状结构造成材料表面畸变,使得表面氧原子逸出并形成大量氧缺陷。在此基础上,利用缺陷结构带来的大量悬键和表面配位不饱和原子,实现了 PML-Cu在BOB表面的有效包覆,最终得到PBOB。
通过HRTEM对比观察了BOB和PBOB的形态(图1b, c)。BOB表面较窄的原子间距为0.274 nm,对应于(2 0 0)或(0 2 0)晶面,而较宽的原子间距则延伸至0.289 nm(与平行于管壁的各向异性晶格拉伸应变相关)。由于PML-Cu包覆了BOB,PBOB表面为无序结构。利用HAADF-STEM(图1d-g)在PBOB观察到距离约为2 nm的高密度分散Cu原子。此外,STEM-EDS图像(图1h)也表明了Bi、O、Br和Cu元素在PBOB中均匀分布。
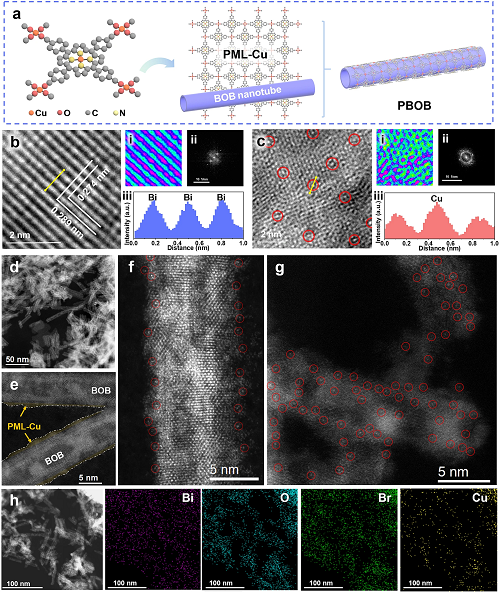
图1.(a)PBOB的合成示意图;(b)BOB和(c)PBOB的HR-TEM(i 是区域伪彩图,ii 是晶格的FFT图,iii 是黄色箭头对应的强度曲线);(d-g)PBOB的HAADF-STEM和(h)STEM-EDS图像。
II PBOB的表界面双极化作用
图2a和b所示为材料的Bi 4f和O 1s XPS谱图。结果显示在PBOB形成后,Bi的特征峰向高结合能方向移动,而氧空位相关峰和PML-Cu中的端基氧信号都向较低结合能方向移动。与此同时,PBOB的EPR(图2c)结果也表明BOB中氧空位浓度的降低,说明PML-Cu有效占据了BOB的表面空位。在材料的Br XPS光谱中未见变化,表明PML-Cu与BOB中[Bi₁₂O₁₇]层的界面重构没有影响卤素层。利用紫外光电子能谱(UPS)计算了材料的功函数(Φ),从而研究其界面电荷迁移。BOB的Φ低于PML-Cu,说明PBOB中内建电场引发的界面极化现象将促进BOB中光生电子流向PML-Cu。利用EPR测试揭示材料表面Cu原子的信息(图2f)。除BOB外,PML-Cu和PBOB都显示出明显的Cu2⁺信号,相较于PML-Cu的PBOB信号变化表明其容纳自旋电子能力及卟啉单元间偶极相互作用的增强。理论计算从原子水平分析了PBOB表面的电子分布情况。如图2g和h所示,电子在Cu原子位点富集,并在卟啉分子骨架中形成耗尽区。PBOB中Cu原子位点电子密度的局部增强表明PBOB的表面极化作用将进一步诱导PML-Cu中电子向Cu原子的转移。基于上述结果,给出了PBOB的表面界面双极化作用示意图(图2i)。具体来说,这是一个多步极化过程,首先BOB和PML间的界面极化能够将电子转移到PBOB表面,而表面PML中的第二阶段极化将促使电子继而转移到Cu位点。
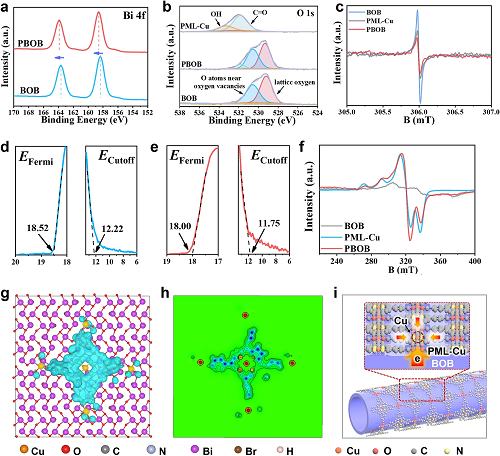
图2.材料的(a)Bi 4f,(b)O 1s XPS和(c)EPR光谱;(d)BOB和(e)PML-Cu的UPS光谱;(f)材料的EPR光谱;DFT计算:PBOB的(g)电荷差密度图像及(h)ELF;(i)PBOB的表界面双极化示意图。
通过紫外-可见漫反射光谱(图3a)分析了材料的光吸收能力。构成PBOB后,材料的Soret峰的偏移及吸光特性的变化表明PBOB中BOB与PML-Cu的界面重构导致了卟啉基元的电荷重布。材料的稳态荧光光谱图(图3b)中,位于420-540 nm处的强荧光信号主要由BOB产生,PBOB的极化作用导致BOB中光生电子的快速转移,进而导致其荧光信号大幅淬灭。瞬态荧光光谱图(图3c)揭示了光生电荷在PBOB的有效空间分离及其显著延长的载流子寿命。通过多种自由基的产生电位及不同材料的产生浓度变化(图3d-f)推测了PBOB的能带结构示意图(图3g)。在光照射下,BOB的CB中高浓度光生电子向PML-Cu迁移,而界面势垒会抑制电子的反向迁移,从而导致光生电子在PML-Cu中富集。通常,BOB的VB主要由Br和O原子相关能级贡献。在PBOB中,卤素层在空间上相对独立于PML-Cu,导致大部分空穴保留在BOB中。最终,PBOB实现了光生电荷的有效分离并可为光催化CO₂还原提供更多的光生电子。
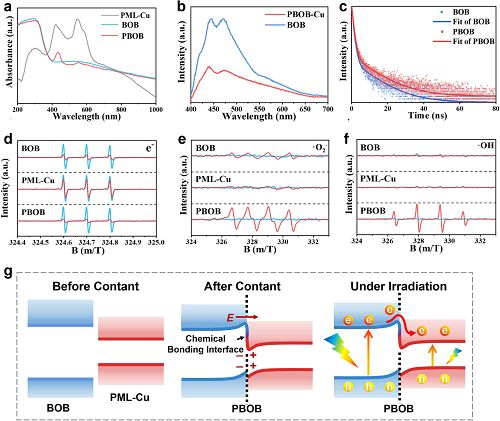
图3.(a)材料的紫外可见漫反射光谱;(b)稳态荧光光谱和(c)瞬态荧光光谱;材料的(d)e-、(e)·O₂-和(f)·OH的ESR光谱;(g)PBOB的能带结构示意图。
III 材料光催化CO₂还原性能研究
在纯水条件下对催化剂的光催化还原CO₂性能进行了评估。如图4a所示,经过5 h反应周期后,PBOB材料光催化CO₂还原为CO的性能为584.3 μmol g⁻1,分别是BOB性能(74.6 μmol g⁻1)的7.83倍,PML-Cu(27.8 μmol g⁻1)性能的21.01倍。条件控制实验(图4b)验证了光和CO₂是反应进行的必要条件。经多次循环测试后(图4c),PBOB仍能保持较好的活性。在与其他先进的光催化剂相比(图4d)后发现PBOB仍具有竞争潜力。此外,通过同位素示踪测试分析了产物的碳来源,结果表明PBOB的光还原作用可有效地将CO₂转化为CO。
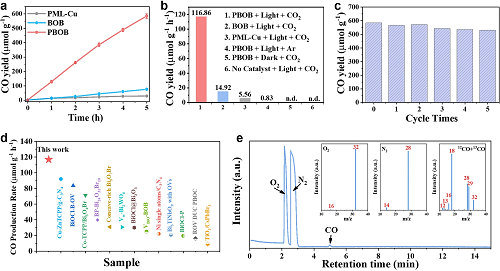
图4.材料的(a)光催化CO₂还原制CO活性图;(b)不同条件下材料的CO产率;(c)PBOB的循环稳定性测试;(d)不同光催化剂的性能比较图;(e)PBOB上12CO₂和13CO₂光还原过程的GC-MS图。
通过线性扫描伏安测试(图5a)评估了PBOB的CO₂还原和析氢反应的竞争关系。当在反应溶液中通入CO₂时,PBOB的电流密度明显增大,验证了PBOB更趋向于发生CO₂还原反应。在光照下,PBOB的电流强度进一步增强,表明光能有效地推动反应进行。利用原位傅立叶变换红外光谱(图5b, c)监测了PBOB表面CO₂的光还原过程。在光照下,能够观察到关键中间体*COOH的持续产生。在反应后期,CO与*COOH的信号比不断增强,说明PBOB的中间体向产物快速转化。采用时间分辨原位MCT-SEIRAS FTIR对催化剂表面连续扫描(图5d, e)。在无光照条件下通入CO₂后,在1300-1660 cm-1范围内出现持续增长的信号表明CO₂在材料表面建立有效化学吸附。光照后,深蓝色区域的形成说明吸附态CO₂的快速消耗,同时红色信号的持续激增表明光照引发CO₂高效还原为CO,验证了CO₂在PBOB表面的快速吸附-扩散-传质过程。
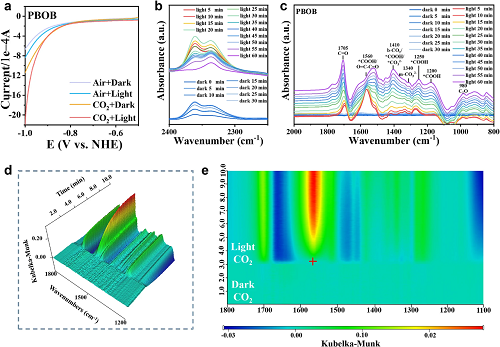
图5.(a)PBOB在不同气氛下的LSV曲线;(b, c)CO₂还原过程的原位傅立叶变换红外光谱图;PBOB的(d)时间分辨的原位MCT-SEIRAS傅立叶变换红外光谱和(e)二维等高线伪彩图。
理论计算研究结果(图6a)表明通过表界面双极化策略构建的PBOB显著降低了大部分反应步骤所需的活化能垒。引入的PML-Cu大大促进了CO₂的吸附(从-0.42到-3.19 eV),并将*COOH形成和关键步骤CO解吸的活化能分别降低了1.27和3.69 eV。各个反应步骤之间活化能势垒差异的减小使得PBOB的CO₂还原过程可以更加顺利地进行。利用差分电荷(图6b、c)探索了临界步骤中CO₂和CO在材料表面的电子传递差异。在CO₂吸附过程中,PBOB中Cu位点转移到CO₂的电子主要富集在C原子。然而,BOB中从Bi原子转移到C原子的电子通过C=O键进一步向O原子扩散,这种分子内电荷转移导致C=O键的键能更强,断键所需的势垒更高,不利于向中间体的转化。在CO解吸时,CO分子中的O原子与BOB表面的Bi位点有更强的亲和力,而CO则通过Cu原子和C原子与PBOB相互作用,减小了CO在PBOB表面的解离能。这些结果与材料的吉布斯自由能变化趋势一致。
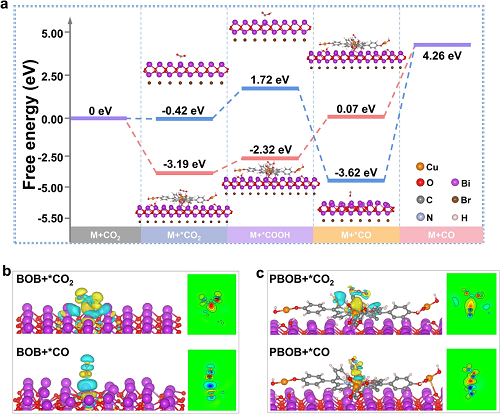
图6.(a)CO₂在BOB和PBOB上还原为CO的吉布斯自由能计算图;(b)BOB和(c)PBOB上CO₂和CO的差分电荷图。
IV 总结
为解决光催化剂活性位点不足和界面电荷转移缓慢的难题,我们开发了应力诱导材料表面界面双极化策略。以Cu-TCPP为基底构建了PML-Cu,并通过应力工程将PML-Cu与BOB组装为PBOB光催化剂。Bi-O键界面在BOB和PML-Cu之间形成了内建电场,驱动电子从BOB的CB转移到PML-Cu的CB,并抑制其反向迁移。PML-Cu的表面极化进一步促进了电子在Cu原子中的聚集。PML-Cu的引入赋予了PBOB表面高密度分散的Cu活性位点,极大地促进了材料对CO₂的吸附和活化以及对CO的解吸。实验表明,PBOB的光催化CO₂还原制CO性能分别是BOB的7.83倍和PML-Cu的20.01倍。这项工作证明了多步极化策略和活性位点密度调控在杂化材料应用中的巨大潜力。
作者简介

本文通讯作者
二维材料设计、能源光催化技术、二氧化碳资源化利用、环境污染物控制技术。

本文通讯作者
铋系材料的设计及其在光催化CO₂转化方面的研究。

本文通讯作者
物质结构及其动力学性质、新型半导体材料与器件。发展了多尺度计算模型及机器学习方法,联合发展超快激光等实验方法研究极端条件下物质的物态物性。

本文通讯作者
主要从事应用化学、精细化工、能源化工、食品化工、材料化工以及化工安全与应急管理技术及产品研发。
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 佘远斌院士等:应力诱导表面界面双极化构建高密度活性位点,提升光催化CO₂还原能力