Anion-Diluent Decoupled Solvation Chemistry in Ionic Liquid-Based Localized High-Concentration Electrolytes Toward High-Voltage Lithium Metal Batteries
Guangye Wu, Haifeng Tu, Zhicheng Wang*, Yiwen Gao, Peng Ding, Yi Yang, Lingwang Liu, Suwan Lu, Farwa Mushtaq, Guochao Sun, Hexiang Chen, Haiyang Zhang, Jiangyan Xue, Jingjing Xu, Hong Li, Xiaodong Wu*
Nano-Micro Letters (2026)18: 396
https://doi.org/10.1007/s40820-026-02242-4
本文亮点
1. 解放FSI⁻,构筑CIP:弱稀释剂-阴离子相互作用使FSI⁻与稀释剂分子解耦,促进离子液体基局部高浓度电解液中形成接触离子对(CIP)主导的溶剂化结构。CIP主导的溶剂化鞘具有更高的本征氧化稳定性,有利于更快的Li⁺脱溶剂化和界面电荷转移动力学。
2. 循环稳定,安全可靠: 基于上述溶剂化调控机制设计的TFE-LHCE电解液,使4.3 V Li||NCM523电池实现600次稳定循环,4.5 V Li||NCM811电池实现200次稳定循环,且本征不可燃,2.6 Ah软包电池能够通过针刺测试。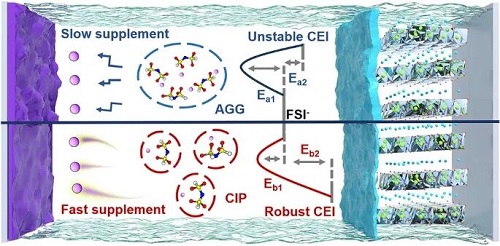
研究背景
高能量密度锂金属电池被视为突破现有锂离子电池能量密度瓶颈的重要方向。然而,这一体系在实际应用中面临多重挑战。锂金属负极具有极低的还原电位,与电解液之间热力学不稳定。与此同时,高镍正极在高电压(≥4.5 V)工况下表面氧化活性显著增强,加剧了电解液的氧化分解以及正极材料衰退。更为棘手的是,常规碳酸酯类电解液高度可燃,一旦发生内短路,极易触发热失控。
离子液体因其物理化学性质稳定、宽的电化学窗口以及本征不燃特性,被认为是高安全电解液的理想载体。但离子液体本身黏度较高、成本昂贵,直接使用难以满足实际电池对离子传输动力学和经济效益的要求。局部高浓度电解液策略能很好的解决该问题,稀释剂与离子液体的搭配,其中离子液体提供了稳定的界面化学和高安全性,稀释剂则弥补了其动力学短板并降低了成本。然而,稀释剂长期以来被简单视为惰性降黏剂,忽视其与体系内其他分子的弱相互作用,事实上稀释剂无论是在非电场作用的体相下还是电池实际工作中的电场作用下的界面相,均对溶剂化结构和界面性质甚至电池性能均存在影响。因此,深入探究稀释剂在离子液体基电解液中的真实角色,对于理性设计高性能、高安全电解液具有重要意义。
内容简介
基于上述认知空白,中国科学院苏州纳米所吴晓东/天目湖先进储能技术研究院王志诚等人以两种结构相似但性质略有差异的氢氟醚稀释剂TFE和TFEE为模型,系统探究了稀释剂与FSI⁻阴离子之间的弱相互作用对锂离子溶剂化结构的间接调控机制。研究发现,稀释剂并不直接配位锂离子,而是通过与阴离子相互作用重塑了溶剂化结构。当稀释剂与FSI⁻作用过强时(如TFEE),部分负电荷从阴离子转移至稀释剂,削弱了FSI⁻与Li⁺的配位能力,迫使体系形成以聚集体(AGG)为主的溶剂化网络。这种结构尺寸庞大、电子富集,不仅阻碍离子传输,还易在高压正极侧发生氧化分解,以及负极侧疏松的锂沉积。反之,当稀释剂与FSI⁻作用恰到好处时(如TFE),可实现阴离子-稀释剂解耦,诱导形成以接触离子对(CIP)为主导的溶剂化结构。其具有三重优势:一是低的脱溶剂化能垒,高的离子电导率;二是氧化稳定性强,在正极界面形成薄而致密的CEI;三是有利于锂金属负极表面致密、无枝晶的沉积。这一溶剂化调控策略,为高电压锂金属电池的电解液设计提供了新思路。
图文导读
I 稀释剂-阴离子竞争配位决定CIP与AGG主导溶剂化结构
图1从分子层面揭示了稀释剂与FSI⁻阴离子之间的弱相互作用对溶剂化结构的调控作用。理论计算表明,TFEE与FSI⁻的结合能更高、静电势更大、电荷转移量更多,说明TFEE对阴离子的亲和力强于TFE。这种差异直接反映在宏观相行为上,三元相图显示TFEE体系互溶区更宽,而TFE体系更容易发生相分离。进一步分子动力学模拟结果表示,在TFE-LHCE中Li⁺与FSI⁻的配位数更高,接触离子对(CIP)占比42.2%、整体呈CIP主导;相比之下,TFEE-LHCE中AGG占比升至45.1%、转为AGG主导。这是由于TFEE过强的阴离子-稀释剂相互作用削弱了FSI⁻与Li⁺的配位能力,迫使体系形成更大的阴离子网络。最后,HOMO能级计算显示,CIP结构的HOMO值(-0.23075)低于AGG(-0.07229),意味着CIP主导的电解液具有更强的抗氧化能力,这对高压正极界面的稳定性至关重要。
图1. 稀释剂与FSI⁻阴离子之间的弱相互作用调控溶剂化结构证据及作用机制。
II 溶剂化结构验证和动力学性能差异
图2从谱学和电化学角度进一步验证了两种溶剂化结构的差异。首先,红外光谱显示,加入稀释剂后FSI⁻在1170 cm⁻¹的特征峰发生蓝移,且TFEE的位移更显著,表明其与FSI⁻的相互作用更强。拉曼光谱定量分析了溶剂化组分:TFE-LHCE中CIP占比约50%、AGG约25%,而TFEE-LHCE中CIP约33%、AGG约41%。广角X射线散射也显示,TFEE体系中的FSI⁻团簇尺寸更大,实验数据印证了其更易形成AGG结构。
在动力学方面,TFE-LHCE的脱溶剂化能明显低于TFEE-LHCE,均方位移曲线和离子电导率实测值均表明TFE-LHCE中Li⁺迁移更快。原位弛豫时间分布分析进一步确认,TFE-LHCE在电池中的界面电荷转移阻抗始终更低。线性扫描伏安曲线显示,TFE-LHCE具有更高的氧化起始电位,说明其抗氧化能力更强。示意图总结了CIP和AGG在离子传输与氧化稳定性上的本质差异。最后,燃烧测试表明,两种离子液体基LHCE均完全不燃,而常规碳酸酯电解液遇火即燃,凸显了该体系的本征安全性。
图2. 光谱学与电化学分析证实CIP主导的TFE-LHCE具有更低的脱溶剂化能、更快的Li⁺传输动力学、更高的氧化稳定性。
III CIP动力学优势, 实现高Li||Cu库仑效率、锂负极界面致密无枝晶锂沉积
进一步系统评估了两种电解液在锂负极侧的界面兼容性与沉积行为。首先,Aurbach法测得的库仑效率显示,TFE-LHCE体系平均CE为98.84%,高于TFEE-LHCE的97.24%。长循环Li||Cu电池测试进一步表明,TFE-LHCE可稳定循环超过600圈,而TFEE-LHCE在约200圈后出现效率波动,400圈左右失效。Li||Li对称电池中,TFE-LHCE可稳定运行2000小时,显著强于TFEE-LHCE。
飞行时间二次离子质谱分析显示,TFE-LHCE形成的SEI在溅射过程中信号逐渐减弱,说明SEI更薄;反之TFEE-LHCE的信号随溅射增强,表明反应层更厚。SEM直观呈现了沉积形貌的差异,TFE-LHCE中锂沉积呈致密块状,厚度约14 μm,表面光滑明亮;TFEE-LHCE中锂沉积呈枝晶状,厚度达30 μm。以上结果表明:CIP主导结构如何通过小尺寸团簇促进快速脱溶剂化和均匀沉积,而AGG主导结构则因大网络增加传输阻力,导致枝晶生长与体积膨胀。
图3. CIP主导的TFE-LHCE在锂负极界面实现高库仑效率、致密无枝晶沉积及超长循环寿命。
IV 缺电子CIP抗氧化作用构筑良好正极界面
图4对比了两种电解液在高镍NCM811正极表面形成的CEI形貌与厚度。FIB-SEM截面图显示,原始NCM811颗粒完整光滑。经TFE-LHCE循环200圈后,二次颗粒出现轻微微裂纹,但表面覆盖均匀的分解产物;而TFEE-LHCE体系则呈现严重的晶间裂纹,分解产物堆积更厚。这是由于AGG主导的TFEE-LHCE在高压下氧化分解更为剧烈,富电子结构加速了FSI⁻的分解。
TEM进一步量化了CEI厚度。TFE-LHCE形成的CEI仅约3 nm,而TFEE-LHCE的CEI厚度达8 nm。较厚的CEI层不仅增加界面阻抗,还会持续消耗电解液并加速正极材料的结构退化。XPS等高线图显示,TFEE-LHCE体系中LiF和有机物种的信号随溅射时间呈现更明显的垂直分布,印证了其更严重的氧化分解。相比之下,CIP主导的TFE-LHCE形成的薄而致密的CEI层有效抑制了副反应,保持了正极的结构完整性,这为其在高电压下的长循环稳定性奠定了基础。
图4. CIP主导的电解液在正极表面形成薄而致密的CEI层,且有效抑制晶间裂纹与电解液氧化。
V 全电池长循环性能与针刺安全验证
图5展示了TFE-LHCE在扣式电池和软包电池中的电化学性能。采用高负载NCM523正极的Li||NCM523电池中,TFE-LHCE稳定循环600次,容量保持率70%;而TFEE-LHCE在约380次后容量快速衰减。当采用NCM811正极、截止电压提升至4.5 V时,TFE-LHCE在0.5 C下循环200次后容量保持率仍达88%,TFEE-LHCE则从早期即出现明显衰退。差分容量曲线进一步证实,TFE-LHCE体系中NCM811的三个相变峰在循环过程中保持稳定,而TFEE-LHCE出现峰位偏移和强度衰减,表明正极结构退化严重。
进一步地,Ah级 Li||NCM83软包电池稳定循环40次,容量保持率95%。此外,满电状态下的针刺测试显示,软包电池被针刺贯穿后未发生起火、爆炸或冒烟,充分验证了离子液体基LHCE的本征安全性。雷达图对比显示,TFE-LHCE在循环寿命、电压耐受、负极兼容性和安全性等关键指标上优势明显。以上结果综合表明,基于阴离子-稀释剂解耦策略设计的TFE-LHCE,在高电压锂金属电池中兼具优异的电化学性能和安全性。
图5. 全电池实现高压长循环稳定,软包电池通过针刺安全测试。
VI 总结
本研究系统揭示了非溶剂化稀释剂通过调控与FSI⁻阴离子的弱相互作用间接决定锂离子溶剂化结构的微观机制,并据此提出了“阴离子-稀释剂解耦”的设计策略。核心发现表明稀释剂与阴离子作用过强会导致大的电荷转移,削弱FSI⁻配位能力,迫使一个Li⁺和多个FSI⁻阴离子配位,导致体系形成AGG主导的溶剂化网络,这种结构尺寸大、电子富集,既阻碍离子传输又易在高压下氧化分解;而当二者作用较小,则可实现解耦,促进单一Li⁺和单一FSI⁻配位,进而诱导CIP主导的溶剂化结构。作为一种结构小巧、缺电子溶剂化结构,不仅具有良好的动力学性能,还兼备良好的抗氧化性能。
基于该策略设计的TFE-LHCE电解液具备本征不可燃的优势,Ah级软包电池通过针刺测试,未发生起火或爆炸。性能方面,在4.3 V Li||NCM523扣式电池中实现600次稳定循环,容量保持率70%;在4.5 V Li||NCM811电池中实现200次稳定循环,容量保持率88%。该工作从分子层面建立了稀释剂-阴离子相互与溶剂化结构之间的关系,为离子液体基局部高浓度电解液的理性设计提供了理论依据。
作者简介


关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc.),包括微纳米材料与结构的合成、表征、性能及其在能源、催化、环境、传感、人工智能、电磁波吸收与屏蔽、健康监测、生物医药等领域的应用研究及高水平综述。期刊已被SCI、EI、PubMed、SCOPUS等数据库收录,2025 JCR IF=38.5,学科排名Q1区前1.5%。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
期刊网址: https://springer.com/40820
投稿网址:https://mc03.manuscriptcentral.com/nmlett
E-mail: editorial_office@nmlett.org
Tel: 86-21-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 中科院苏州纳米所吴晓东&天目湖先进储能院王志诚团队:稀释剂—阴离子解耦离子液体电解液,实现高压高安全锂金属电池