研究背景
超级电容器具有高输出功率和长寿命等优点,弥补了传统电容器与可充电电池之间的差距,在各种商业应用中得到了广泛的应用。在混合超级电容器中,金属锌由于其高比容量、低氧化还原电位、低成本以及丰富的储量成为一种有吸引力的负极材料。然而,水系电解液窄的电化学窗口会导致严重的副反应(比如析氢和电偶腐蚀),进而缩短循环寿命。电解液工程是提高水系超级电容器能量密度和循环寿命的有效策略。比如,通过溶解高浓度的盐制备“盐包水”电解液,可以有效拓宽电化学窗口。而很多电解液添加剂也可以通过在锌负极表面形成吸附层或者改变电解液的溶剂化结构来抑制锌负极的腐蚀和析氢。然而,这些策略不可避免地增加了电解液的密度和粘度,从而降低了其能量密度和倍率性能。因此,需要设计更合理的电解液体系实现这些特性间的平衡。
Dilute Aqueous‑Aprotic Electrolyte Towards Robust Zn‑Ion Hybrid Supercapacitor with High Operation Voltage and Long Lifespan
Shuilin Wu, Yibing Yang, Mingzi Sun, Tian Zhang, Shaozhuan Huang, Daohong Zhang, Bolong Huang,* Pengfei Wang & Wenjun Zhang*
Nano-Micro Letters (2024)16: 161
https://doi.org/10.1007/s40820-024-01372-x
本文亮点
1. 一种新的低盐浓度水/非质子电解液(0.5 mol Zn(CF₃SO₃)₂ + 1 mol LiTFSI)表现出扩展的电化学窗口,既可以稳定Zn金属负极,又可以提高Zn离子混合超级电容器的工作电压。
2. 由乙腈和LiTFSI诱导的电解质的配位壳不仅可以抑制Zn的腐蚀和析氢反应,还可以促进阴极稳定性和离子迁移。
3. 基于开发的电解质的Zn离子混合超级电容器可以在宽的温度范围内以及在0-2.2 V的电压范围内稳定运行,具有超过120,000次的超长循环寿命。
内容简介
水系电解液的电化学窗口较窄,导致锌金属负极发生严重的副反应,缩短了其使用寿命,也制约了锌离子混合超级电容器的工作电压和和能量密度。使用“盐包水”电解液可以有效地扩大其电化学窗口,但同时也会导致高成本、低离子电导率以及窄的温度范围,损害了锌离子混合超级电容器的电化学性能。因此,迫切需要设计一种新的电解液来平衡这些特性,从而实现高性能的锌离子混合超级电容器。香港城市大学张文军与香港理工大学黄勃龙等人开发了一种用于锌离子混合超级电容器的稀的水/乙腈电解液(0.5 mol Zn(CF₃SO₃)₂ + 1 mol LiTFSI-H₂O/AN),同时具有宽的电化学窗口、良好的离子电导率和宽的温度相容性。该电解液中,乙腈和TFSI⁻阴离子调控了水合壳层和氢键,使用该电解液的锌离子混合超级电容器具有高达2.2 V的高工作电压和超过120,000次的长循环寿命。
图文导读
I Zn负极在0.5 m Zn(CF₃SO₃)₂+1 m LiTFSI H₂O/AN电解液中的电化学性能
图1a所示为Zn负极在不同电解液中的Tafel曲线。其中,较高的峰值电位有利于抑制析氢过程,因此在H₂O/AN电解液中锌沉积/溶解过程的电位较高,表明析氢副反应被有效抑制。图1b所示为原电池中Zn负极随时间变化的腐蚀电流曲线,H₂O/AN电解液中电流密度的波动明显降低,意味着电偶腐蚀的降低。图1c显示在10 mA cm⁻2的电流密度下,在0.5 m +1 m H₂O/AN电解液中仍保持97.3%的高的库伦效率,而在0.5 m +1 m H₂O电解液中库伦效率则迅速下降。图1d的对称电池的循环曲线显示,随着电流密度的增加,使用0.5 m +1 m H₂O/AN电解液的Zn-Zn对称电池的的电压迟滞略有增加。此外,图1e和1f分别通过Zn-Cu和Zn-Zn电池验证了Zn负极在0.5 m +1 m H₂O/AN电解液中的长循环性能。其中,Zn-Cu电池在3000次循环中保持97.3%的高库伦效率。此外,Zn-Zn对称电池也展现出了超过1000次的稳定循环,而是用0.5 m +1 m H₂O电解液时则迅速发生短路。
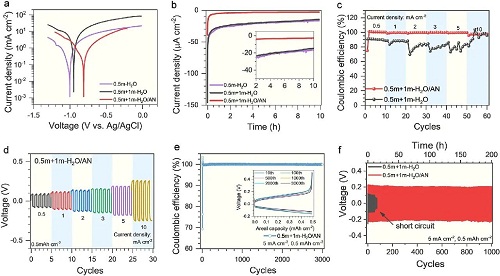
图1. 锌金属负极在0.5 m Zn(CF₃SO₃)₂+1 m LiTFSI H₂O/AN电解液中的稳定性:在0.5 m + 1 m-H₂O/AN电解液和参考的水系电解液中(a)Zn沉积/剥离的Tafel曲线;(b)Zn-Ti原电池电流密度随时间的变化曲线;(c)不同电流密度下Zn-Cu电池的库伦效率;(d)不同电流密度下Zn-Zn对称电池的电压曲线;(e)Zn-Cu电池在0.5 m + 1 m-H₂O/AN电解液中的库伦效率;(f)不同电解液中Zn-Zn对称电池的循环稳定性。
II 锌离子混合超级电容器在0.5 m Zn(CF₃SO₃)₂+1 m LiTFSI H₂O/AN电解液中的电化学性能
使用多孔石墨烯(aMEGO)和锌箔分别作为正极和负极,组装了锌离子混合超级电容器。图2a所示的恒流充放电曲线显示在2.2 V以下时,Zn-aMEGO混合超级电容器的充放电曲线保持了线性和对称性,而在2.2 V以上时,充放电曲线出现平台期。图2b利用CV曲线评估了Zn-aMEGO混合超级电容器的倍率性能,扫描速率从5 mV s⁻1增加到100 mV s⁻1时,CV曲线保持了准矩形的规则形状。图3c同样证实在电流密度从0.5增加到20 A g⁻1时,电容保持率仍高达64%。图2d,2e结果显示,使用0.5 m +1 m H₂O/AN电解液的Zn-aMEGO混合超级电容器可以在0-2.2 V的高工作电压范围内可以以88%的保持率稳定循环超过120000次。
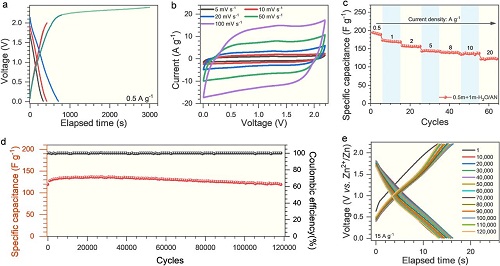
图2. Zn-aMEGO混合超级电容器在0.5 m + 1 m-H₂O/AN电解液中的电化学性能:(a)恒流充放电曲线;(b)不同扫描速率下的循环伏安曲线;(c)倍率性能;(d)15 A g⁻1电流密度下的循环稳定性;(e)不同循环圈数的恒流充放电曲线。
进一步研究了H₂O/AN电解液的低温性能,评估其在恶劣条件下的实用性。图3a所示在-30℃下,Zn-Cu电池随着电流密度增加,观察到了高的库伦效率和轻微增加的电压迟滞。同样,图3b也证实在低温下,Zn-Zn对称电池在增大电流密度时也出现略问增加但稳定的电压迟滞。图3c显示,基于0.5 m +1 m H₂O/AN电解液的Zn-Cu电池在-30℃下具有高的库伦效率(99.4%)和良好的循环稳定性(500次循环)。这些结果表明,即使在恶劣的温度条件下,在0.5 m +1 m H₂O/AN电解液中也保持了高可逆性和快速的Zn沉积/剥离。图3d显示使用0.5 m +1 m H₂O/AN电解液的Zn-aMEGO超级电容器在宽的温度范围内保持了良好的电化学性能,CV曲线畸变很小。相比之下,使用0.5 m +1 m H₂O电解液时,CV曲线在低温时变得扭曲,并且积分面积极具减小(图3e)。图3f显示,使用0.5 m +1 m H₂O/AN电解液的Zn-aMEGO超级电容器在-30℃下可以稳定循环超过2000次。
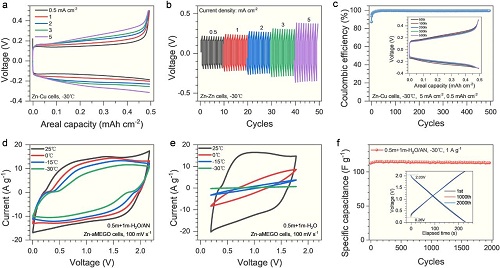
图3. 在0.5 m + 1 m-H₂O/AN电解液中的低温电化学性能:(a)Zn-Cu电池在不同电流密度下的充放电曲线;(b)Zn-Zn电池在不同电流密度下的电压曲线;(c)Zn-Cu电池在5 A g⁻1电流密度下的库伦效率;(d)0.5 m + 1 m-H₂O/AN电解液和(e)0.5 m + 1 m-H₂O电解液中Zn-aMEGO混合超级电容器在不同温度下的循环伏安曲线;(f)Zn-aMEGO超级电容器在0.5 m + 1 m-H₂O/AN电解液中的电容保持率。
III Zn(CF₃SO₃)₂ + LiTFSI H₂O/AN电解液的溶剂化结构
通过分子动力学(MD)模拟,对不同浓度的H₂O/AN电解液进行了密度泛函理论(DFT)计算。图4a所示,0.5 m +1 m H₂O电解液中费米能级附近的成键轨道和反键轨道分别在Zn(CF₃SO₃)₂和TFSI⁻附近。Zn(CF₃SO₃)₂与水分子之间较弱的轨道耦合表明,原有的溶剂化结构不稳定,可能导致副反应,降低整体性能。图4b的0.5 m +1 m H₂O/AN电解液中引入AN,AN的s和p轨道对成键和反键轨道都有很强的贡献,增强了轨道耦合。然而,当LiTFSI的浓度增加到3 m时,成键轨道和反键轨道主要由LiTFSI主导,水和AN的贡献减弱,增加了电子转移的能垒(图4c)。随着LiTFSI浓度进一步增加到5 m,水分子在成键轨道中的贡献变得更强,这增加了HER的可能性(图4d)。同时,轨道耦合进一步减弱,导致电子转移阻碍增加,这与过量LiTFSI引起的实验表征阻抗增大是一致的。图4e的PDOS揭示了0.5 m +1 m H₂O电解液的电子结构,H₂O的s和p轨道与LiTFSI和Zn(CF₃SO₃)₂均有明显的重叠,支持了溶剂化结构的形成,但3 eV的带隙限制了整体的导电性。图4f所示为0.5 m +1 m-H₂O/AN电解液中,AN的相互作用导致Zn-3d轨道发生轻微的下移。并且H₂O与LiTFSI和Zn(CF₃SO₃)₂的相互作用明显减弱,说明AN的加入改变了溶剂化结构。随着LiTFSI浓度的增加,观察到Zn-3d轨道的显著下移,表明水分子与Zn2+和乙腈分子之间的相互作用进一步受到抑制,同时能隙也随之增大,导致了高的电子转移势垒(图4g,4h)。图4i的RDFs证实,0.5 m +1 m H₂O电解液中的成键贡献主要来自于水分子中的H-O键。而在图4j的0.5 m +1 m H₂O/AN电解液的RDFs中,水分子的峰几乎消失。图4k所示为不同电解液中离子与溶剂分子的相互作用能,0.5 m +1 m H₂O/AN电解液中相互作用能降低,相互作用更强。图4l的均方位移图表明,0.5 m +1 m H₂O/AN电解液中具有最高的扩散。与水系电解液相比,H₂O/AN电解液中的离子相互作用更强,离子和分子的迁移率更高,不仅提高了电解质的导电性,而且提高了超级电容器的工作电压。
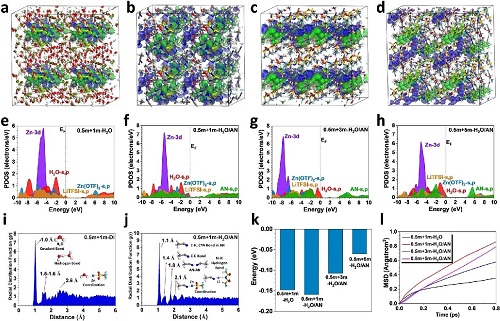
图4.(a)0.5 m + 1 m-H₂O电解液,(b)0.5 m + 1 m-H₂O/AN电解液,(c)0.5 m + 3 m-H₂O/AN电解液和(d)0.5 m + 5 m-H₂O/AN电解液的反键轨道和成键轨道的三位轮廓图,其中蓝色等值面代表成键轨道,绿色等值面代表反键轨道;(e)0.5 m + 1 m-H₂O电解液,(f)0.5 m + 1 m-H₂O/AN电解液,(g)0.5 m + 3 m-H₂O/AN电解液和(h)0.5 m + 5 m-H₂O/AN电解液的PDOS图;(i)0.5 m + 1 m-H₂O电解液和(j)0.5 m + 1 m-H₂O/AN电解液的径向分布函数图;(k)作用能比较图;(l)不同电解液在298K下的均方位移图。
IV 总结
本文设计了一种新型的稀的水/乙腈电解液,用于锌离子混合超级电容器,具有优异的电化学性能。理论计算证实,乙腈分子与Zn阳离子具有较强的相互作用,调控了电解液的溶剂化结构,有利于扩大其在低盐浓度下的电化学窗口。乙腈形成的配位壳层不仅抑制Zn腐蚀和HER,而且提高了整体电化学性能。在稀的0.5 m +1 m-H₂O/AN电解液中,Zn沉积/剥离的平均库仑效率达到97.3%,并且锌离子混合超级电容器具有超长寿命(> 120,000次循环)。此外,得益于水-非质子电解液的低盐浓度,锌离子混合超级电容器即使在-30°C的恶劣条件下也能稳定运行,拓展了其应用场景。本研究为通过调控溶剂化结构来开发新型电解质,实现高能量密度、长寿命的锌离子混合超级电容器提供了新的途径。
作者简介

本文通讯作者
纳米材料、能源材料、固体功能材料和稀土材料的电子态性质,以及在能源材料纳米表界面、多尺度下的能源转换应用等。
▍Email:bhuang@polyu.edu.hk

本文通讯作者香港城市大学 讲席教授
薄膜技术、纳米材料和器件。
▍Email:apwjzh@cityu.edu.hk
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 香港城市大学张文军等:低浓度水系-非质子电解液助力锌离子混合超级电容器具有高工作电压和长寿命