研究背景
实现锂离子电池更安全、更快速的充电对诸如电动汽车等电动工具的推广应用具有十分重要的意义。然而,Li⁺插层的缓慢动力学限制了其快速嵌入石墨层中。在使用较高倍率充/放电时,石墨负极具有较大的极化和较小的嵌锂容量,同时还伴随着一些副反应,如锂金属的沉积、较厚固体电解质(SEI)膜的形成和焦耳热的产生等。如何加快Li⁺在石墨负极中的嵌入反应动力学是实现高性能石墨负极用于快充锂离子电池的关键。本文利用冷冻透射电子显微镜(cryo-TEM)和电位弛豫技术(PRT)等测试分析手段,解析了Li⁺在界面、石墨层间和电极内部的扩散,揭示了在大电流密度嵌锂过程中石墨结构的演变规律,并探索其与反应动力学和电化学性能之间的内在联系。
本文亮点
1. 由于缓慢反应动力学的限制,快速嵌锂的石墨结构中存在多种“阶”化合物,在宏观和微观上都表现出明显的不均匀性。
2. 在石墨薄电极中,Li⁺在界面处的扩散是限制石墨倍率性能的关键步骤;而在石墨厚电极中,Li⁺在电极内部的扩散不可忽视。
内容简介
全面认识石墨在快速嵌锂过程中的结构演变和速率限制步骤对于加快Li⁺在石墨负极中的嵌入反应动力学至关重要。对此,中国科学院物理研究所/北京凝聚态物理国家研究中心王雪锋特聘研究员和王兆翔研究员等人利用冷冻透射电子显微镜(cryo-TEM)和电位弛豫技术(PRT)等测试分析手段,揭示了Li⁺快速嵌入过程中石墨体相和界面结构的演变规律,并将其与反应动力学和电化学性能相联系。快速嵌入的Li⁺局域地分布在石墨表面,嵌锂石墨的体相结构在宏观和微观上都表现出多种“阶”化合物共存的状态。Li⁺化学扩散系数计算结果表明,粒径为10 μm的石墨至少可以实现6 C倍率下的快速嵌锂。因此,与Li⁺在石墨层间的扩散相比,Li⁺在界面处的扩散是石墨薄电极在大电流密度下嵌锂反应动力学瓶颈,这与电解液和SEI膜性质密切相关。然而,对于石墨厚电极而言,Li⁺在电极内部的扩散不可忽视,结合电极结构设计和电解液改性可以显著提升其倍率性能。
图文导读
I Li⁺嵌入石墨过程
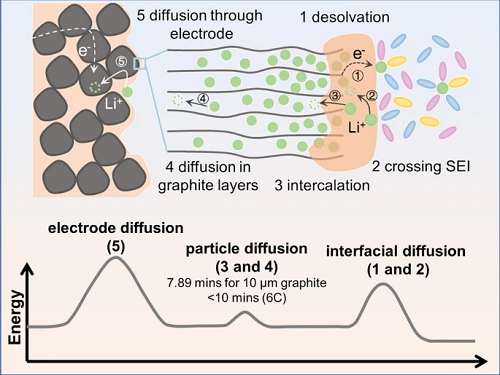
Li⁺在嵌入石墨需要经历以下几个过程(图1):(1)溶剂化Li⁺发生去溶剂化;(2)去溶剂化后的Li⁺通过SEI膜输运;(3)Li⁺嵌入石墨;(4)Li⁺在石墨层间扩散;(5)Li⁺在石墨电极内部扩散。上述过程可以进一步分成三个主要的能量消耗步骤:Li⁺在界面、石墨层间以及石墨电极内部的扩散。
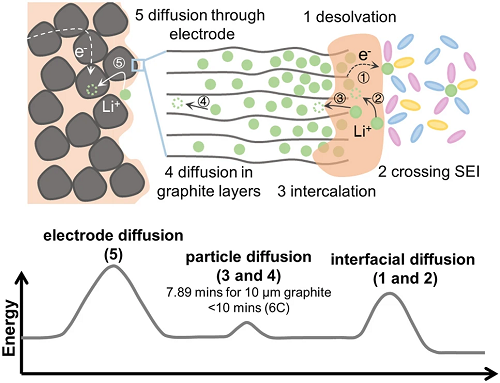
图 1. Li⁺嵌入石墨过程微观图像和相应的能量消耗示意图,包括界面扩散、石墨层间扩散和石墨电极内部扩散。
II 快速Li⁺嵌入/脱出时石墨体相结构的演变
使用不同速率(0.05 C vs. 0.5 C)将相同含量的锂(250 mAh g⁻1)嵌入到石墨中,并细致地对比了嵌锂石墨的宏观和微观结构。XRD表征发现,快速嵌入的Li⁺不均匀地分布在石墨片层中,引起多“阶”共存的现象(图2a-b)。Cryo-TEM结果表明,在微观上,0.05 C嵌锂石墨主要由Ⅱ阶和少量Ⅲ阶相构成(图2c),而0.5 C嵌锂石墨中同时存在Ⅰ-Ⅳ阶相结构,在纳米尺度上呈现明显的结构不均匀性(图2d)。
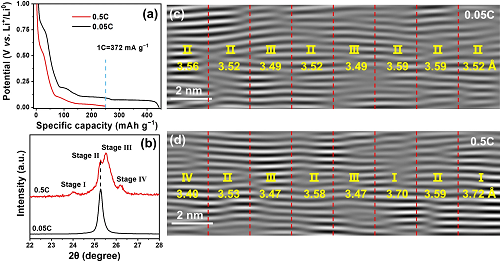
图 2. 石墨在慢速(0.05 C)和快速(0.5 C)嵌锂到相同容量(250 mAh g⁻1)时的结构对比。(a)比容量–电压曲线;(b)XRD谱图;(c-d)iFFT图像。
快速锂化石墨中的局域应力场和Li⁺的面分布结果表明,石墨快速嵌锂过程受到缓慢反应动力学的限制,Li⁺只能嵌入石墨的亚表层,呈现出不均匀的分布,导致可逆容量降低(图3)。围绕上述的三个能量消耗过程对石墨大倍率嵌锂的限制步骤进行讨论。
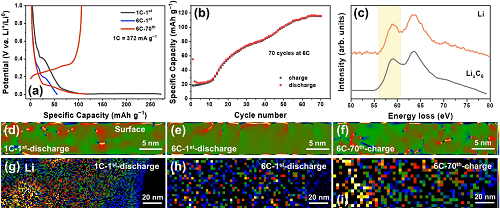
图3. 大倍率嵌锂石墨性能和结构表征。(a)石墨在1 C和6 C循环时的比容量–电压曲线;(b)6 C循环性能;(c)Li的K吸收边的EELS谱线;石墨在1 C和6 C嵌锂后及6 C循环70周后体相内的(d-f)局域应力场和(g-i)LiₓC₆分布。
III Li⁺在石墨层间扩散
采用电位弛豫技术(PRT)测量了Li⁺在不同“阶”嵌锂石墨中的化学扩散系数,并估算在不同颗粒尺寸石墨中,Li⁺从表面扩散到体相内所需时间(图4)。随着Li⁺不断地嵌入,扩散系数先从初始的19.87×10⁻1⁰ cm2 s⁻1下降到2.64×10⁻1⁰ cm2 s⁻1,然后略微上升到3.45×10⁻1⁰ cm2 s⁻1,最后稳定在3.25×10⁻1⁰ cm2 s⁻1。基于嵌锂过程中的最小扩散系数Dmin(2.64×10⁻1⁰ cm2 s⁻1),估算出Li⁺从粒径为d的石墨颗粒表面扩散到体心所需要的时间。对于直径为10 μm的石墨,Li⁺从石墨颗粒表面扩散到体心所需要时间约为7.89 分钟(对应的倍率为7.60 C),小于6 C快充时所需的时间(10分钟)。这表明Li⁺在石墨体相中的扩散足够快,能够满足6 C快速充放电的要求。
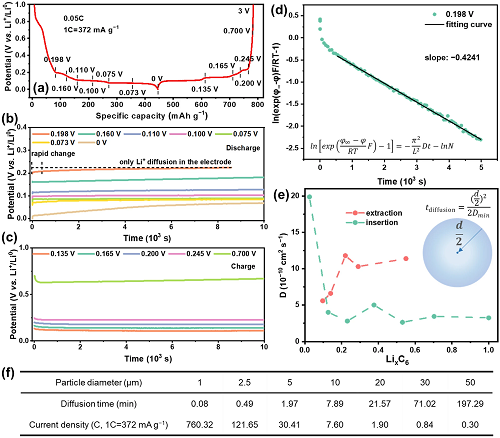
图 4. 采用PRT方法测定Li⁺在石墨体相中的化学扩散系数。(a)石墨在0.05 C嵌锂时的比容量-电压曲线;(b-c)选取图a中标注的不同(b)嵌锂/(c)脱锂状态下石墨的电位弛豫曲线;(d)以图b中嵌锂到0.198 V的石墨为例示范化学扩散系数D的拟合计算过程,对–时间t分布图进行线性拟合,实线表示拟合结果,斜率即;(e)Li⁺在嵌入/脱出过程中嵌锂石墨的化学扩散系数D;(f)根据嵌锂过程中最小的Li⁺化学扩散系数(Dmin=2.64×10⁻1⁰ cm2 s⁻1)估算Li⁺从直径为d的石墨颗粒表面扩散到中心所需时间和对应的电流密度。
IV 界面调控改善石墨倍率性能
界面Li⁺扩散包括溶剂化Li⁺去溶剂化和Li⁺通过SEI膜输运过程。由于SEI膜具有多孔结构,难以严格区分这两个过程,但是它们都与电解液的化学性质密切相关。因此,通过调节电解液组分,如添加少量FEC,有望降低去溶剂化能垒并形成富含LiF的SEI膜,加快Li⁺在SEI膜中的输运动力学,提升石墨负极的倍率性能(图5a-b)。结果表明,FEC的加入可以显著地提高石墨在高倍率下的可逆容量,特别是2 C以上(图5a)。在Li||GR半电池中,使用FEC电解液的石墨负极在4 C循环时比容量为276.2 mAh g⁻1,在6 C循环时比容量可达145.5 mAh g⁻1;而使用不含FEC电解液的石墨负极在4 C时比容量仅有62.0 mAh g⁻1。在GR||LFP全电池中,电解液添加FEC后,在2 mA cm⁻2和6 mA cm⁻2循环时可逆容量分别保持在300.7和70.5 mAh g⁻1。
通过cryo-HRTEM和cryo-EELS细致地比较了SEI膜的纳米结构。结果表明,在LiPF₆-EC/DMC电解液中,石墨表面的SEI膜较薄,只有3–7 nm厚(图5c和e),由无定形的有机组分和Li₂CO₃纳米小晶粒组成。在添加FEC的电解液中,石墨表面附着纳米小颗粒,由无机物小晶粒镶嵌在有机物基底上构成的,其中内层LiF和外层Li₂O构成核–壳结构(图5d)。通过EELS元素面分布证明了上述核-壳结构:F元素的EELS信号主要集中在小颗粒中心位置,而O元素在其表面富集(图5f)。将小颗粒内外层的Li和F的K吸收边EELS谱线与Li₂O和LiF的标准谱进行对比,进一步确认了小颗粒是内层含LiF外层含Li₂O的核-壳结构(图5g-h)。这种富含LiF的岛状SEI膜的厚度为44–56 nm,均匀地分布在石墨表面,可以有效地调节界面性质,促进Li⁺在界面处的快速输运。
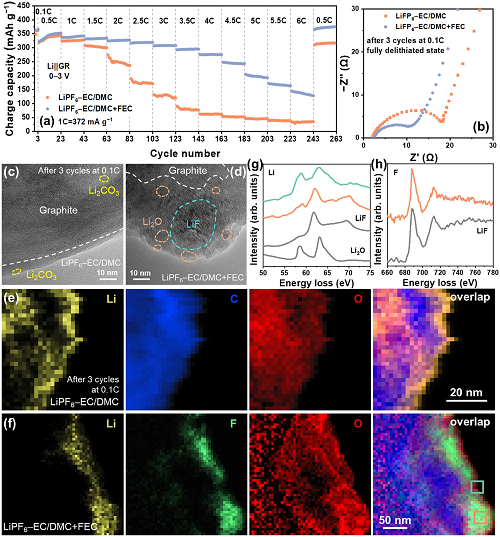
图5. Li||GR电池的电化学性能和界面表征。(a)Li||GR电池的倍率性能;(b)采用三电极装置(其中石墨为工作电极,新鲜的Li箔为对电极和参比电极)测试Li||GR在0.1 C循环3周后的EIS;(c-d)HRTEM图像;(e-f)EELS元素面分析;(g-h)图f中橙色方框和绿色方框区域(g)Li K边和(h)F K边的EELS谱。
V 石墨电极厚度对快速嵌锂动力学的限制
上述对于Li⁺在界面和石墨层间扩散的分析是基于未辊压过的石墨薄电极进行的。然而,当活性物质负载量增加、电极厚度增大和孔隙率减小时,Li⁺在石墨电极内部的扩散不可忽视,甚至可能成为限制石墨负极倍率性能的关键因素(图6)。极片负载量从1.9增加到4.6和6.1 mg cm⁻2时,垂直方向的Li⁺扩散距离从35.52增大到80.34和113.17 μm,高倍率下可逆容量显著降低。对于实用化高负载量低孔隙率的石墨负极,合理设计电极结构,促进Li在电极内部扩散至关重要。同时,相较于传统电解液,使用添加FEC电解液的石墨厚电极表现出更优异的倍率性能,说明电解液调控和SEI膜设计对于石墨厚电极的倍率性能的提升也是必要的。
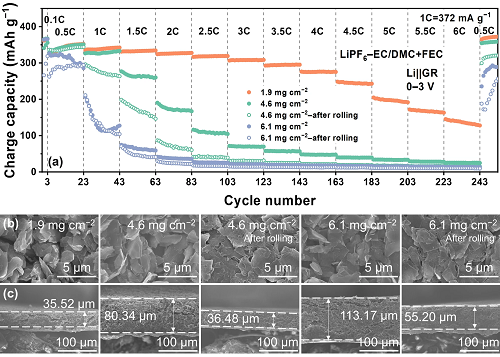
图 6. 不同活性物质面载量和厚度的Li||GR电池倍率性能。
作者简介
 王雪锋
王雪锋本文通讯作者
▍Email:wxf@iphy.ac.cn
 王兆翔
王兆翔本文通讯作者
二次电池材料结构设计、性能预测、材料内部及表面的物理化学过程。
关于我们
 Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 物理所王雪锋&王兆祥等:解析锂电池快充中石墨电极的动力学极限