Nano-Micro Letters (2022)14: 43
https://doi.org/10.1007/s40820-021-00785-2
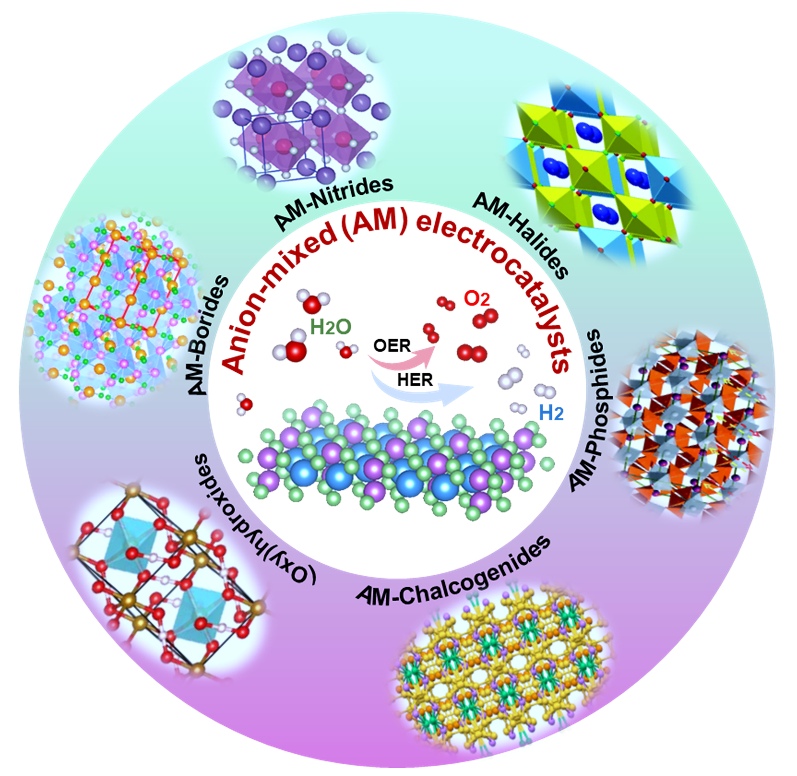
2. 提出并分析了阴离子混合电解水催化剂面临的挑战和未来展望,包括聚阴离子混合策略、阴离子混合无金属催化剂构建、改良的合成策略、先进的原位表征以及在原子水平上对构效关系的解析等。
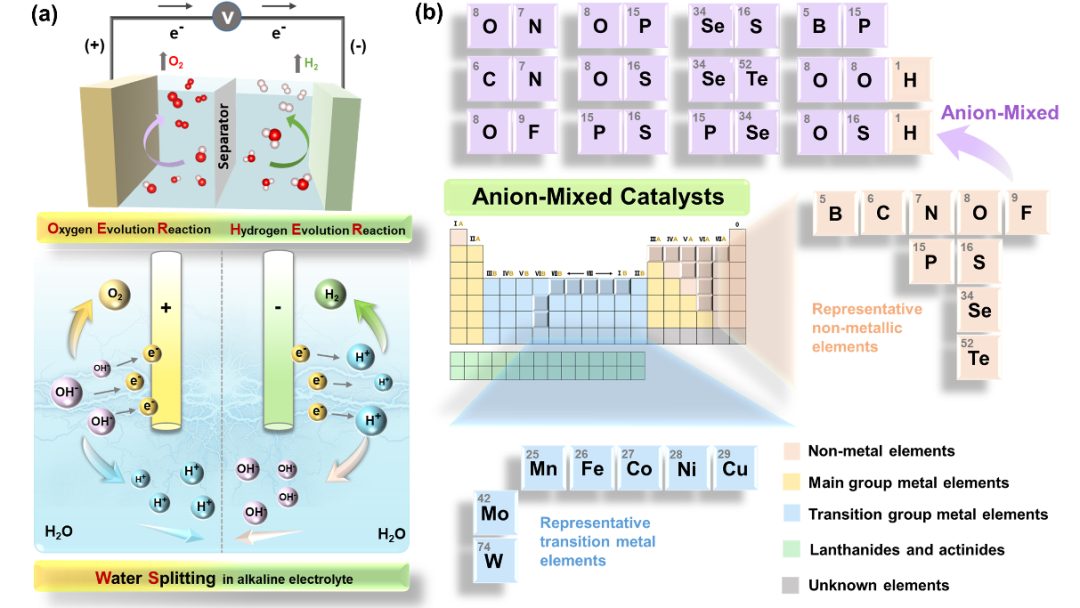
目前,贵金属催化剂的大规模商业化应用受到了严重的阻碍。鉴于此,已经探索了过渡金属(TM)基氧化物、碳化物、氮化物、磷化物、硫属化合物、氢氧化物、卤化物和硼化物。然而,这些材料存在催化性能低、电子导电性差、钝化和溶解等问题,阻碍了它们的实际应用。因此,迫切需要先进的改性策略来提升催化剂的活性和稳定性,其中,阴离子(取代、调制等)策略显示出巨大的潜力。特别是,阴离子混合策略被认为是一种非常有前途的催化剂开发途径。
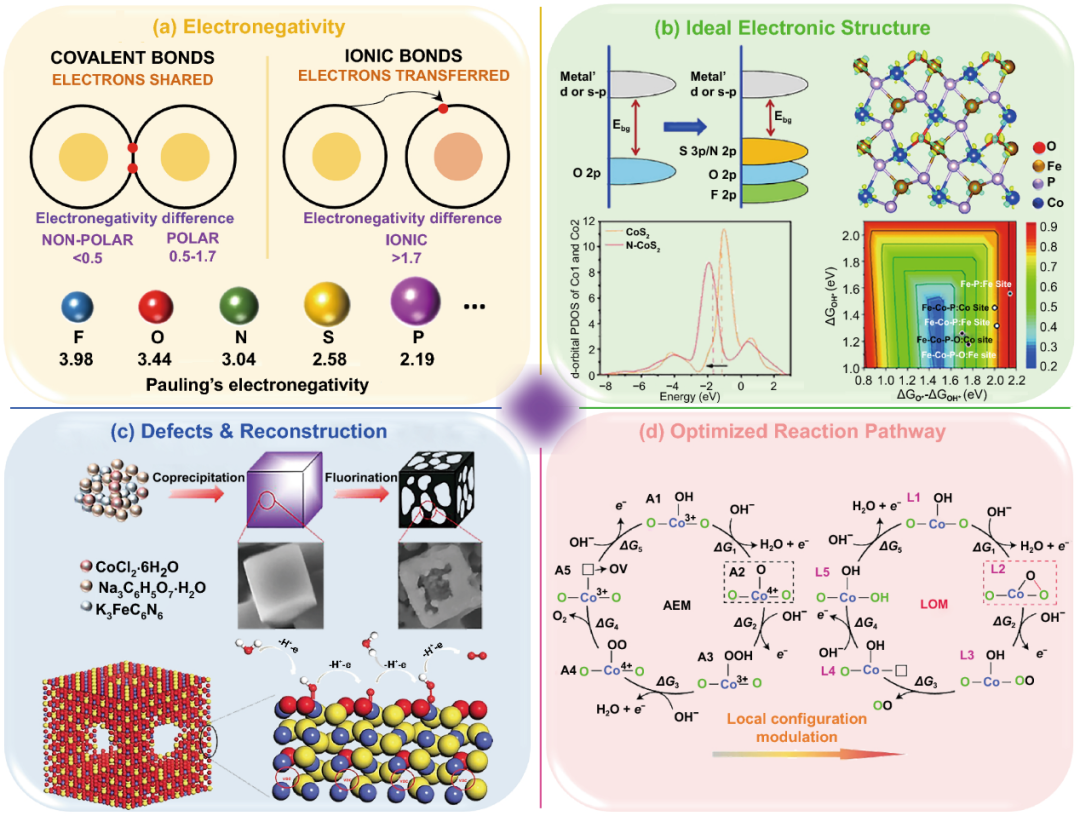
II 阴离子混合策略对催化剂性能提升的四个方面
3.1 混合氮化物
3.1.1 氮氧化物
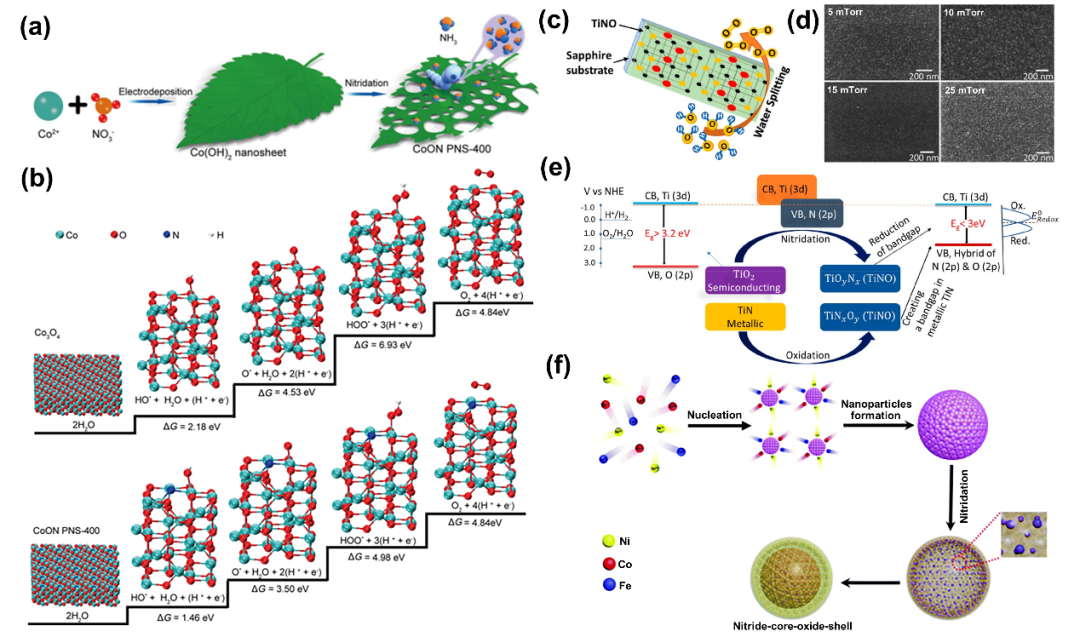
氮(N)与氧化物的结合开辟了调节物理和化学性质的简便途径。由于N的电负性较弱,比O更易极化,因此M-N键在本质上比M-O键更具共价性。此外,N的离子半径比O大,N在氧化物中的取代会增加键合距离。因此,调节TM基化合物中的N比可以改变它们的结构,改善电荷转移过程,并提高电催化活性。
3.1.2 碳氮化物
碳氮化物由于其理想的电子结构而被认为是有效且有前途的电催化剂,其费米能级附近的态密度(DOS)与贵金属的态密度相同。TM和碳或氮原子之间的p-d状态平衡可以通过额外的元素进行调整,这会显著影响其宏观机械性能、微观结构和电子稳定性。
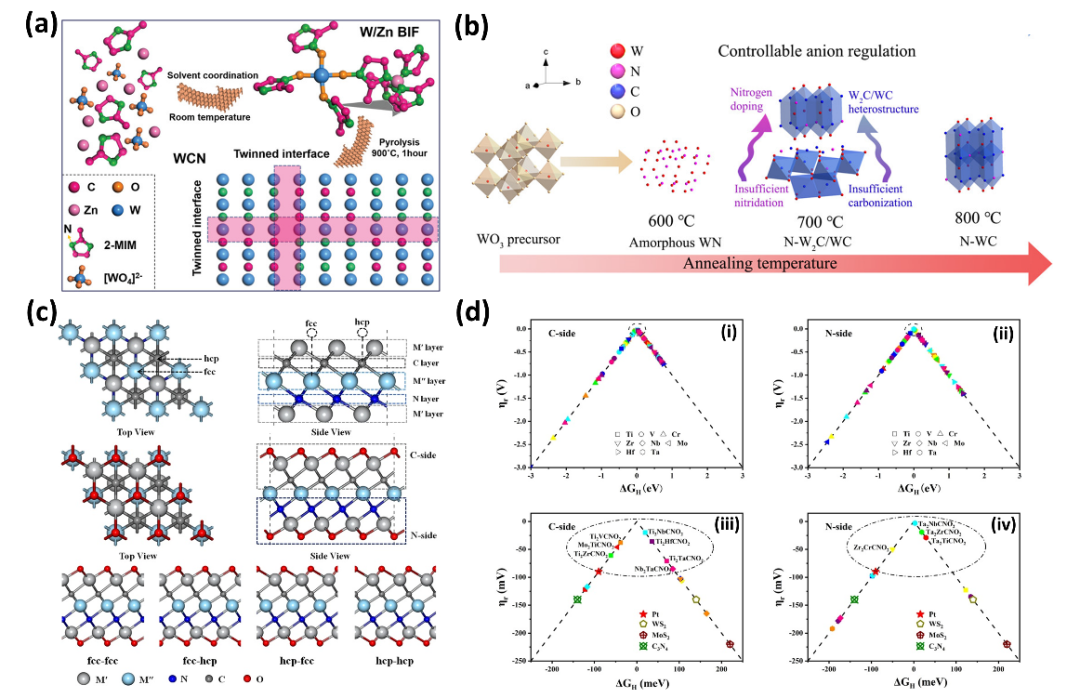
3.2 混合卤化物
3.2.1 氟氧化物
TM氧氟化物(OF)具有独特的电化学催化特性。引入金属-氧(M-O)键可以改善金属-氟化物本质上的弱导电性,因为高共价M-O键可以很好地调节金属-氟(M–F)键的高离子性。F原子都具有很强的电负性,可以很容易地从邻近的金属原子捕获电子,从而形成许多配位不饱和的位置。
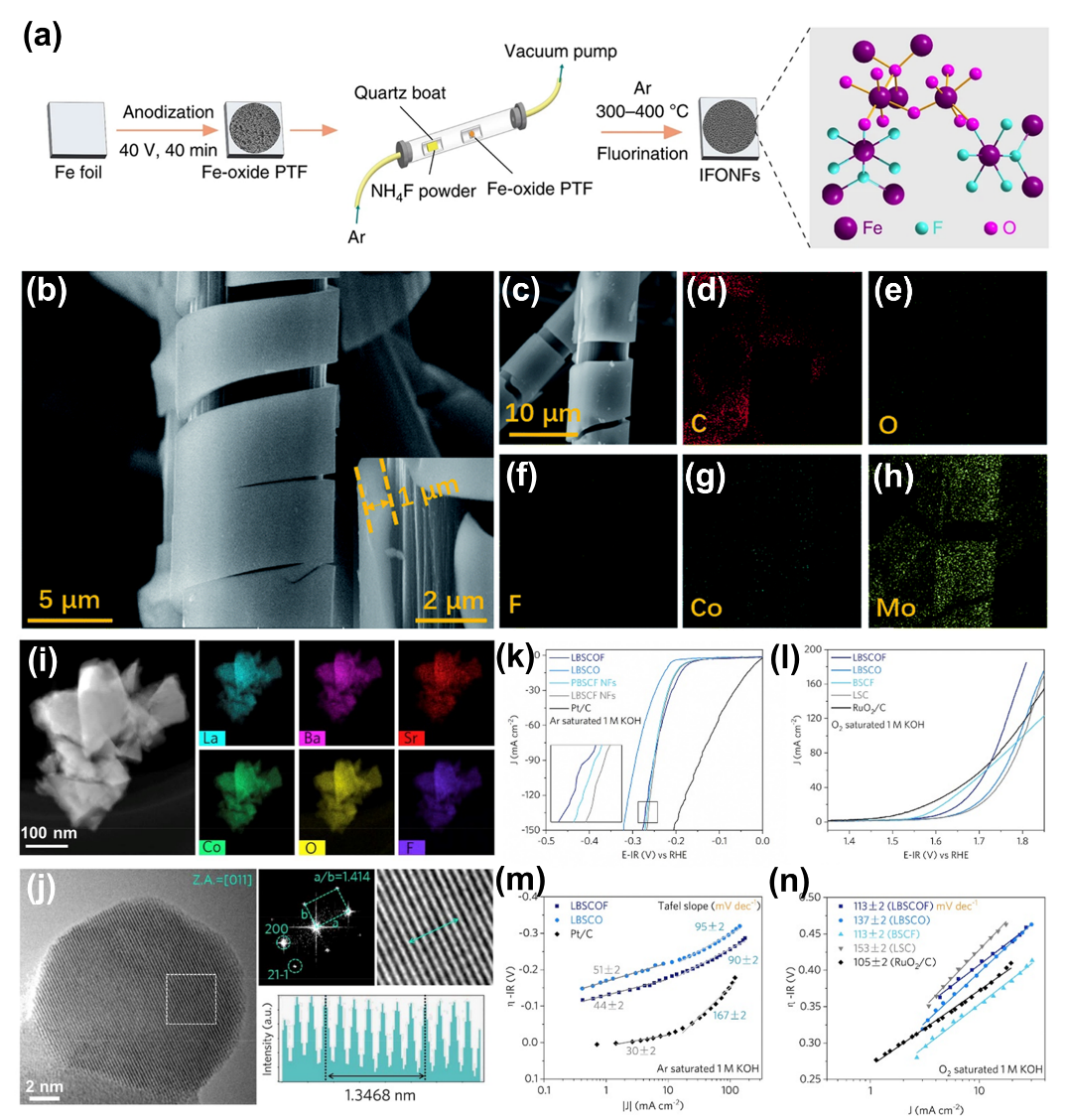
由于金属羟基氟化物(M-(OH)F)键的高度离子性,F阴离子取代被认为是提高金属羟基氧化物催化活性、表面极性和动力学的保证。有趣的是,在主体羟基氧化物晶格中加入F阴离子改善了化学键的极性,从而增强了氢氧化物(OH)阴离子和含氧中间体的吸附能力。
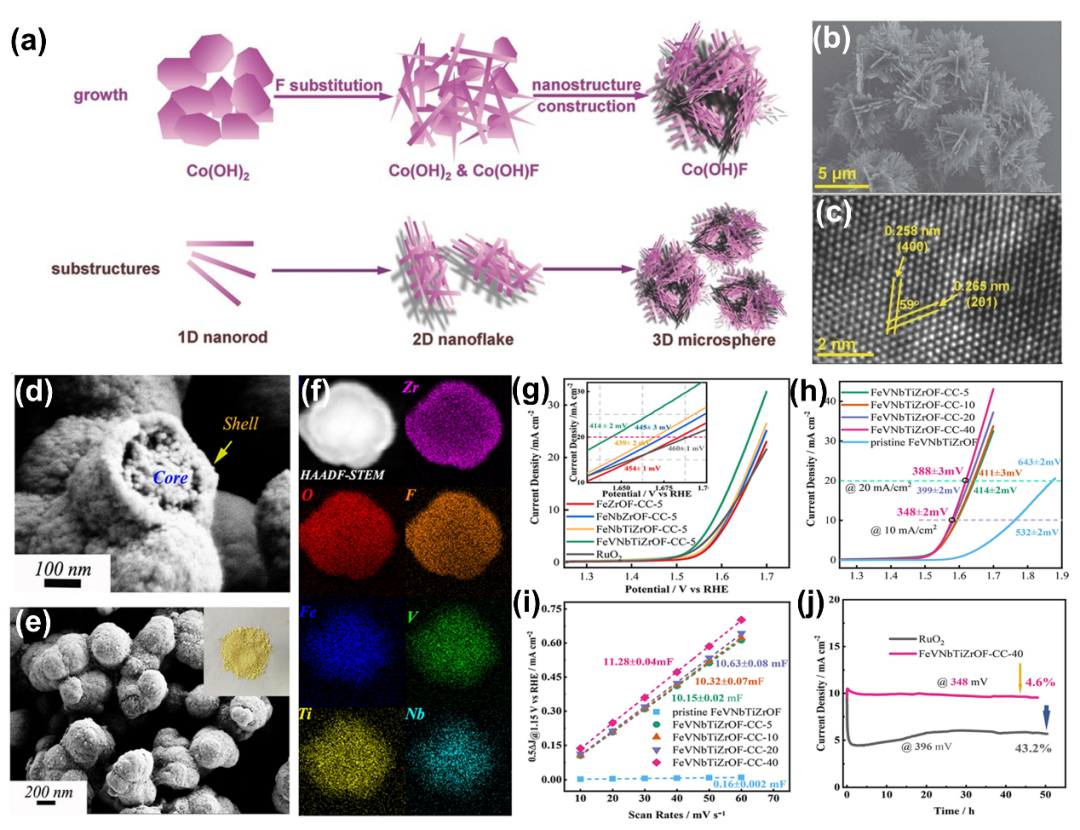
3.3 混合磷化物
3.3.1 氧磷化物
金属磷化物(M-P)中的氧(O)阴离子掺入可以进一步提高电催化剂的固有活性。事实上,由于氧磷化合物中M-P-O金属键的牢固性,它已被证明具有极好的稳定性。此外,氧磷化合物中的金属-P(M-P)和金属-O(M-O)键将有助于调整电子结构,优化中间体的吸附/脱附自由能。磷和氧的相互作用产生了丰富的表面空位/缺陷和较大的活性表面积,有助于提升其催化活性。
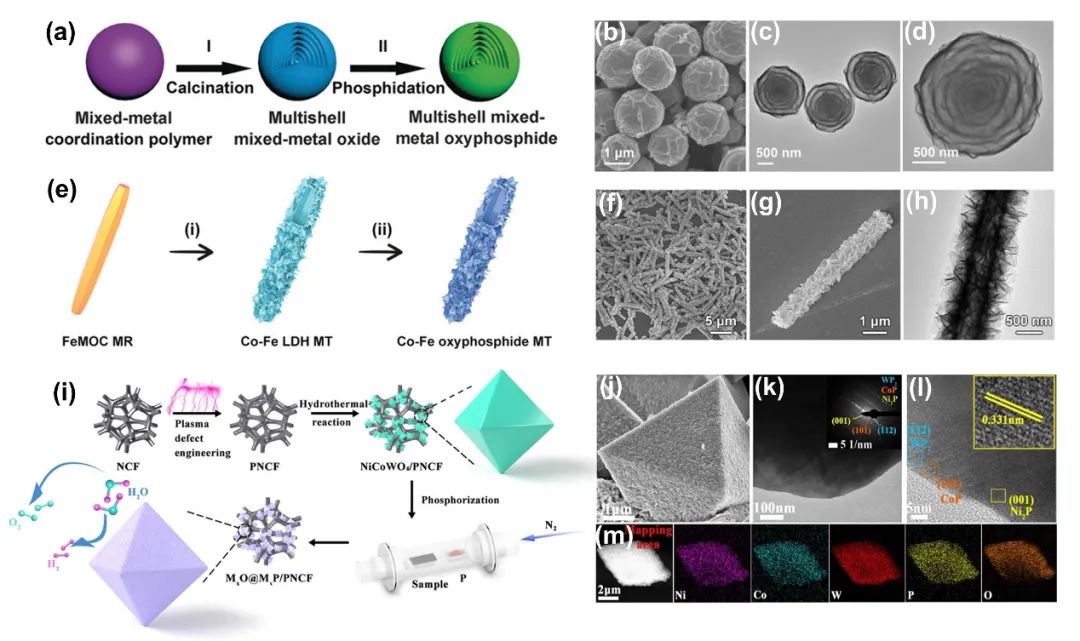
3.3.2 羟基磷酸盐
羟基磷酸盐(HP)是另一种具有代表性的磷氧化物。根据之前的报告,在碱性电解质中,TM-HP通常比相应的TM磷酸盐更耐用。
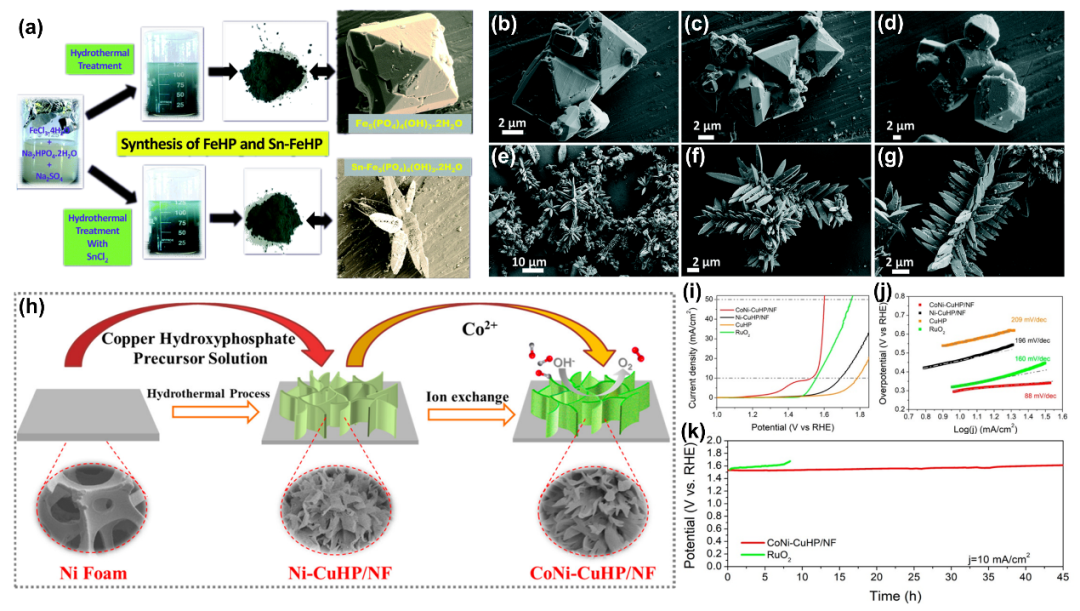
3.4 混合硫属化合物
3.4.1 硫氧化物
氧阴离子通常是“硬”的(非极化),硫阴离子是“软”的(在硫化物中极化)。从周期表的同一VIA群衍生出的O和S很容易混合以修饰化合物。
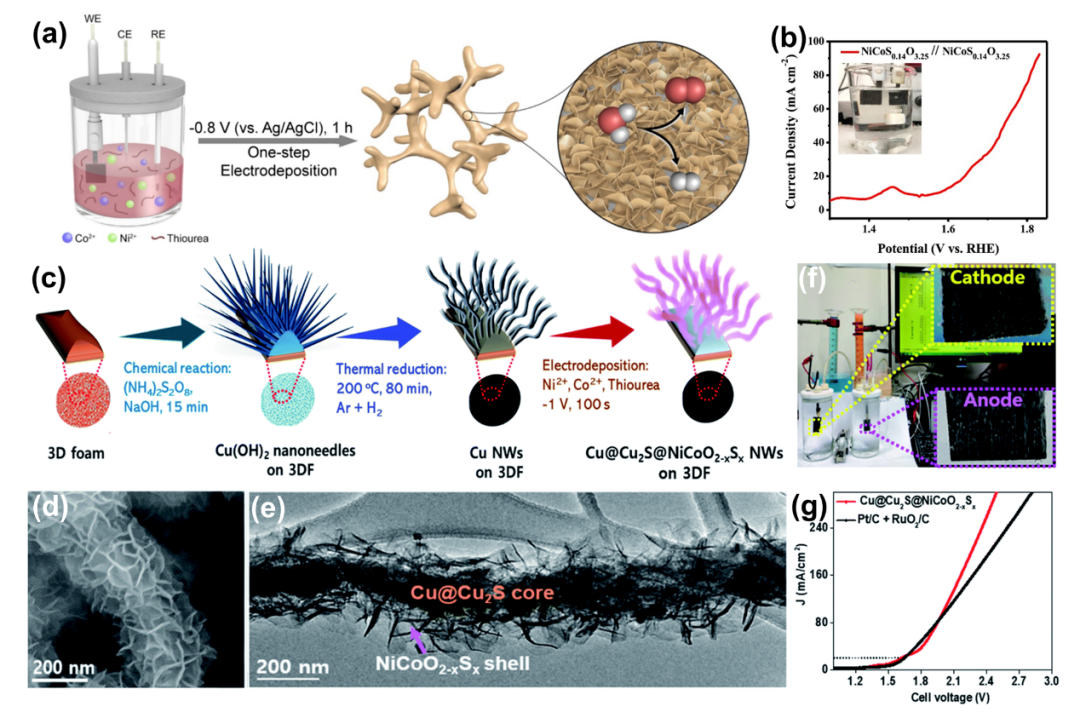
3.4.2 磷硫化物
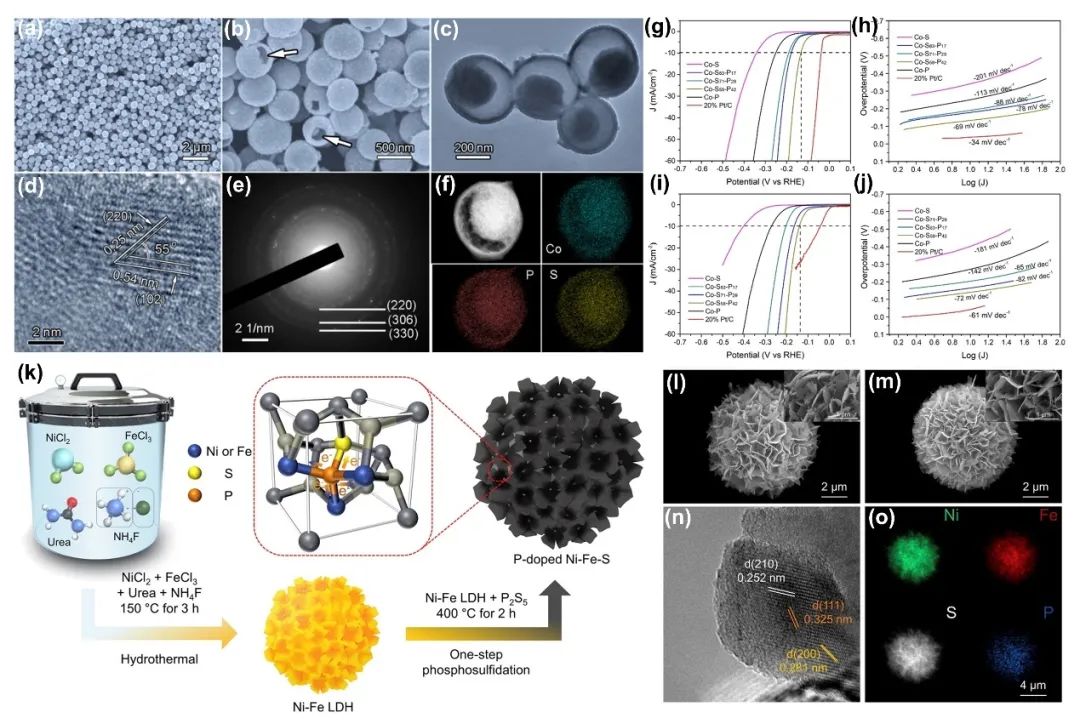
TM磷硫化物(TMPS)表现出优异的催化性能。硫的电催化活性完全取决于硫族配体(X₂²⁻)的电子给予。而P原子增强了X₂²⁻的给电子能力,这促进了金属原子的氧化还原反应。
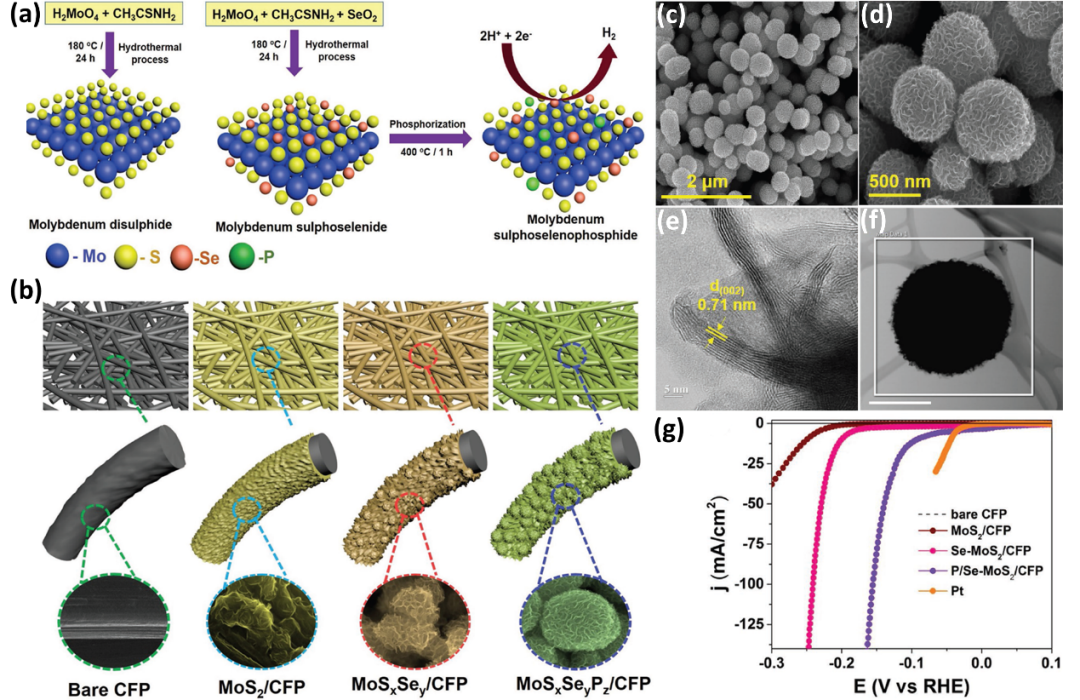
3.4.3/4 硒硫化物/硒碲化物
硒硫化物中控制硫、硒(Se)替代和改变组成比可以改善HER/OER性能。
3.4.5 羟基(硫酸盐、硫化物)
与其他三元/四元TMCs材料一样,TMHydroxy(硫酸盐、硫化物)因其高活性和稳定性而广泛应用于多相催化。DFT计算表明,OH和S配体在NiCo₂(SOH)ₓ表面的协同效应能够调节金属活性中心周围的电子结构及其化学环境(图12d, e)。
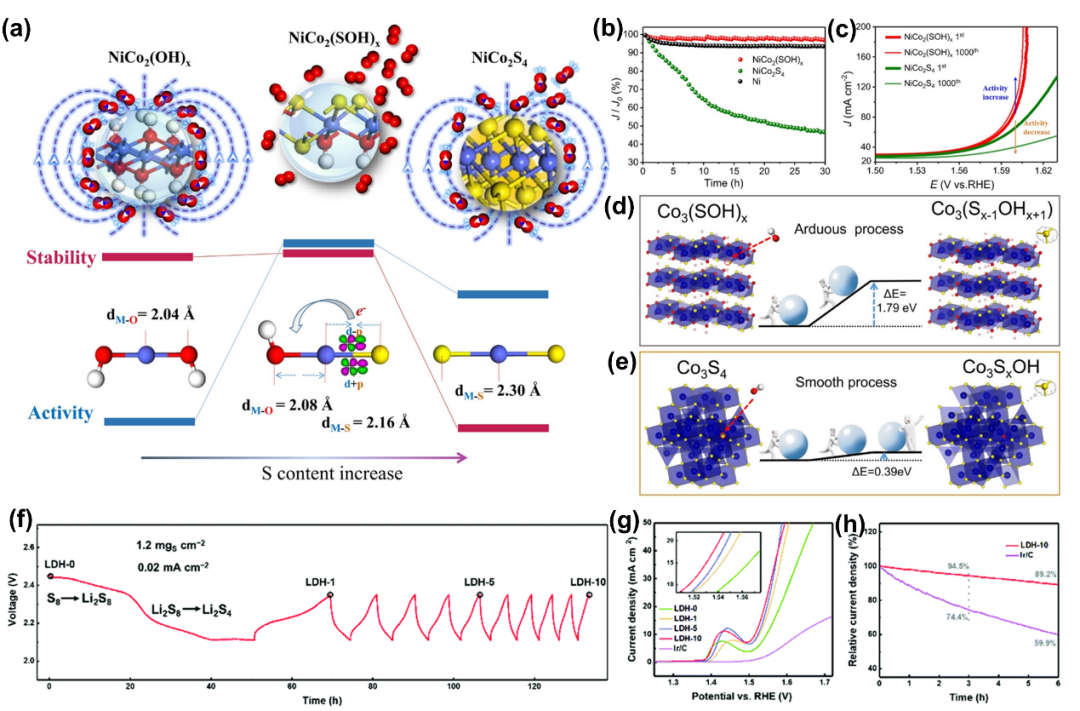
3.4.6 磷硒化物
硒取代磷化物(PSe)将是实现卓越电化学性能的重要步骤。硒掺杂增强了内部磷化物的电子转移,并提供了更多的活性位点。
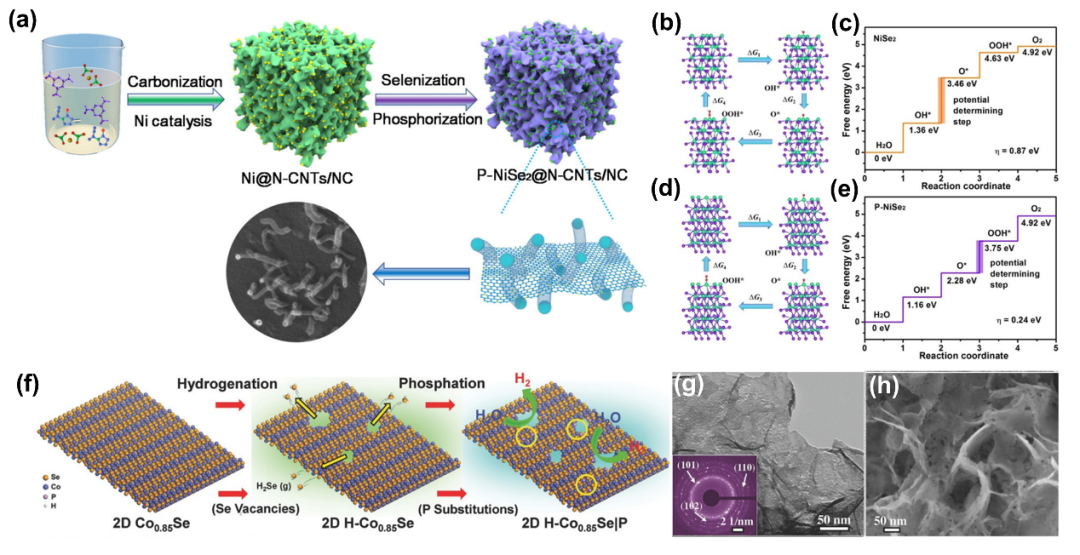
3.5 (氧)氢氧化物
TM-OOH是由TMO₆八面体分层堆叠而成,质子夹在层之间,不同的插层物种可以改变层间距。特别是,镍、铁、钴和锰基(含氧)氢氧化物因其多样性、碱性介质中的大量活性中心、潜在稳定性和低成本而被广泛研究作为高效的水分解催化剂。
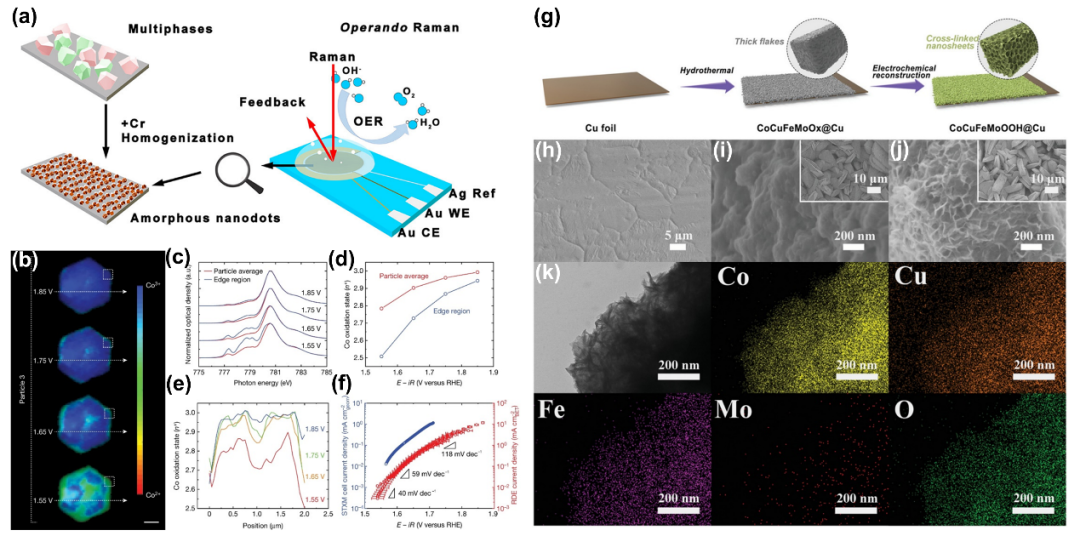
3.6 混合硼化物/硼酸盐
过渡金属硼化物/硼酸盐(TMB)中金属-B键可影响不同位点电子转移性质。例如,硼通过在许多非晶态钴和镍基硼化物中提供电子而起到牺牲作用。从B到金属的反向电子转移导致电子在金属部位富集,防止金属部位氧化,提高其稳定性,同时促进电化学反应。
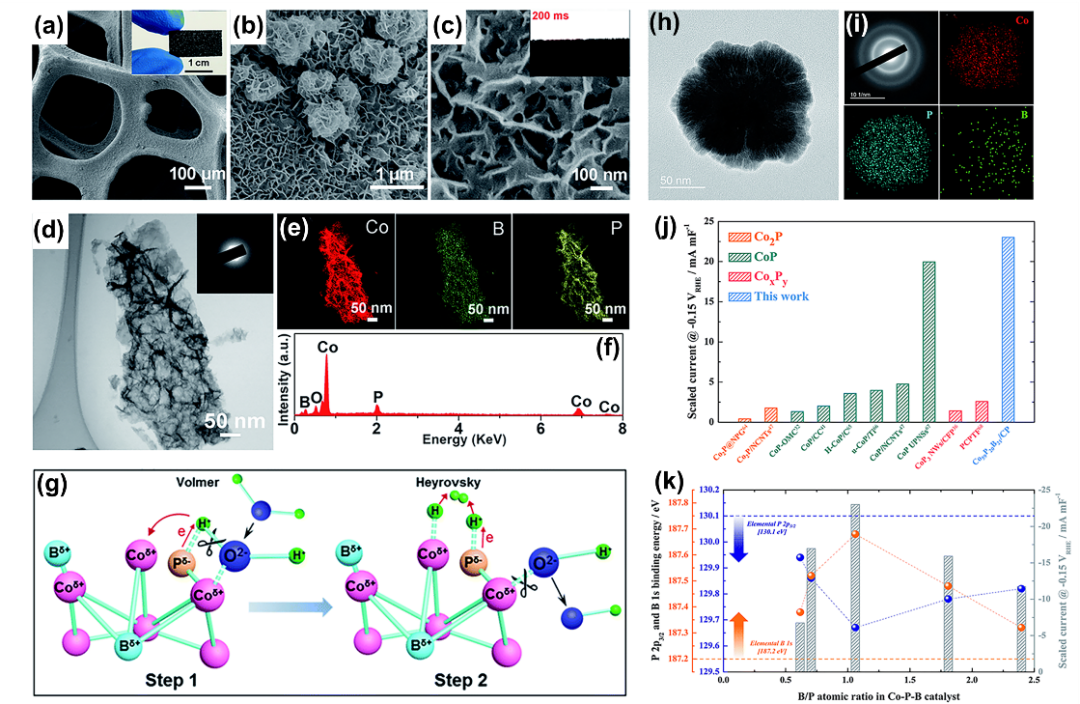
图15. (a-g) 泡沫镍上Co-B-P纳米片的形貌微结构表征及其HER电催化机理示意图。(h-k) Co₅₉P₂₀B₂₁/CP催化剂的微结构、酸性HER性能及其B/P比例-电子结合能对应关系。
IV 总结与未来展望
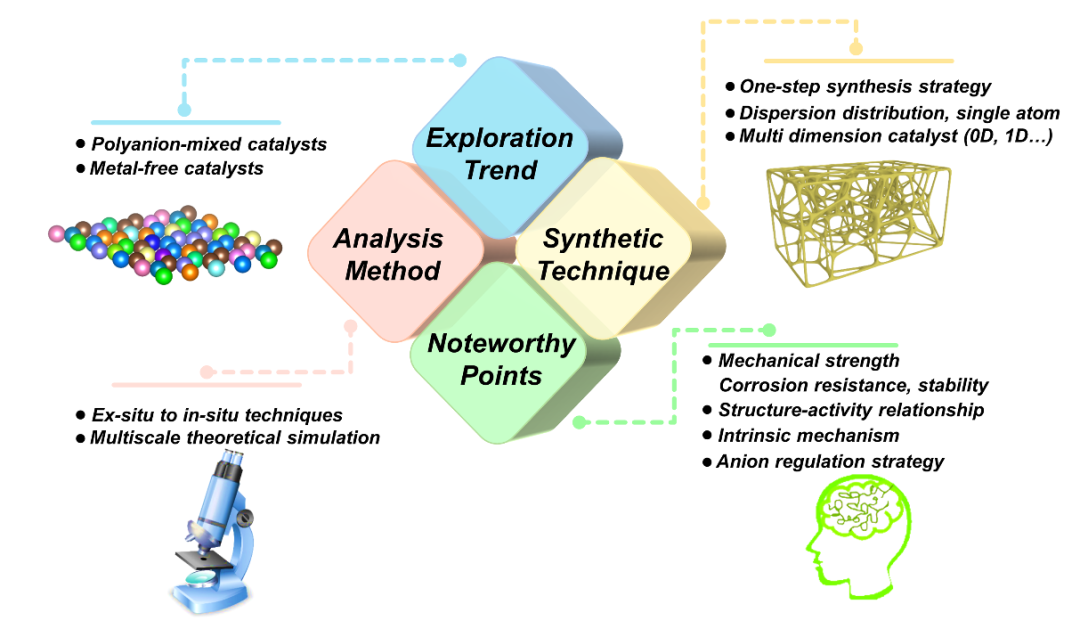
图16. 水电解用混合阴离子催化剂的研究展望。
 戴正飞
戴正飞
本文共同通讯作者
西安交通大学 特聘研究员
能源环境纳米材料与器件。
▍主要研究成果
▍Email: sensdai@mail.xjtu.edu.cn
 Paranthaman Vijayakumar
Paranthaman Vijayakumar
本文共同通讯作者
西安交通大学 助理研究员
主要从事染料敏化太阳能电池、储能和电催化材料研究。
▍Email: kumarphysics89@gmail.com

Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » NML综述 | 阴离子混合水电解纳米催化剂:基础、进展和展望